Número Atual: Janeiro-Março 2017 - Volume 1 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Doenças autoinflamatórias em adultos: abordagem prática ao diagnóstico baseada em um caso clínico
Autoinflammatory diseases in adults: practical approaches to diagnosis based on a clinical case
Leonardo Oliveira Mendonça1; Ricardo Krieger Azzolini2; Andre Franco Silva3; Jorge Kalil4; Alessandra Pontillo5; Fabio Morato Castro6; Myrthes Toledo Barros7
DOI: 10.5935/2526-5393.20170014
1. Médico Imunologista e Alergista da Disciplina de Imunologia Clínica e Alergia do Hospital das Clínicas da Faculdade de Medicina da Universidade de Sao Paulo (HC-FMUSP)
2. Médico Reumatologista da Disciplina de Reumatologia do HC-FMUSP
3. Aluno de Graduaçao da Faculdade de Medicina da Universidade de Sao Paulo
4. Professor Titular da Disciplina de Imunologia Clínica e Alergia da Universidade de Sao Paulo
5. Professora Titular do Instituto de Ciências Biomédicas da Universidade de Sao Paulo
6. Professor Associado da Disciplina de Imunologia Clínica e Alergia da Universidade de Sao Paulo
7. Médica Assistente do Serviço de Imunologia Clínica e Alergia do HC-FMUSP
Endereço para correspondência:
Leonardo Oliveira Mendonça
E-mail: leonardo.oliveira.mendonca@gmail.com
Submetido em: 10/01/2017
Aceito em: 15/02/2017
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
As doenças autoinflamatórias sao doenças inflamatórias raras cujo cerne imunológico baseia-se na imunidade inata. A maioria das doenças autoinflamatórias tem início na idade pediátrica, mas pouco se sabe sobre as doenças que se iniciam na vida adulta. O diagnóstico é feito por exclusao, e, quando possível, com auxílio de técnicas moleculares. Este artigo tem como objetivo relatar um caso de doença autoinflamatória de início na vida adulta e a partir dele estabelecer fluxograma de auxílio ao diagnóstico.
Descritores: Inflamaçao, imunidade inata, doenças hereditárias autoinflamatórias, síndromes periódicas associadas à criopirina, febre familiar do Mediterrâneo, Imunoglobulina D.
INTRODUÇAO
As doenças inflamatórias sistêmicas, inicialmente classificadas como autoimunes, ganharam enorme redefiniçao nas últimas décadas com o aprofundamento do conhecimento imunológico1. Com base no compartimento imunológico envolvido, atualmente, essas doenças sao classificadas em dois grandes grupos: autoinflamatórias e autoimunes. Apesar de parecer apenas definiçoes semânticas, esta diferenciaçao é imprescindível para o diagnóstico correto, direcionamento do tratamento imunológico e prognóstico clínico2.
Como um novo grupo de doenças, as autoinflamatórias tiveram maior reconhecimento no cenário clínico pediátrico desde a descoberta do gene responsável pela Febre Familiar do Mediterrâneo (FFM, OMIM #249100) em meados da década de 903. A FFM, protótipo deste grupo de doenças, apresenta grande prevalência em relaçao às demais doenças autoinflamatórias, acometendo cerca de 100 mil indivíduos em todo o mundo, particularmente os de ascendência do leste do Mediterrâneo (judeus sefárdicos, armênios, turcos e árabes)4. O gene relacionado à FFM é o MEFV (Mediterranean fever gene), situado no braço curto do cromossomo 16, sendo sua herança autossômica recessiva, com várias mutaçoes descritas5 que geram uma alteraçao na proteína pirina, que induz secreçao excessiva de interleucina-1β6.
As manifestaçoes clínicas geralmente ocorrem antes dos 10 anos, com episódios recorrentes de febre (38-40 °C), dor abdominal difusa ou localizada, constipaçao intestinal, artralgia (em grandes articulaçoes), artrite (em joelhos e membros superiores/inferiores) e dor torácica (pleurite e/ou pericardite) que geralmente duram de 1 a 4 dias. Há envolvimento cutâneo caracerizado por lesoes eritematosas, principalmente em membros7. Estas crises podem ser precedidas por mialgia, cefaleias, náuseas e dispneia. Meningite asséptica, esplenomegalia, linfonodomegalia e poliarterite nodosa podem ocorrer. A amiloidose secundária pode ser uma complicaçao grave em longo prazo8,9. Contudo, sua classificaçao e caracterizaçao clínica é bem estabelecida atualmente somente na faixa etária pediátrica10. Em adultos, pouco se sabe sobre essas doenças, seja por fenótipos diferentes ou por manifestaçoes incompletas do esperado, resultando em um atraso do diagnóstico.
Este trabalho tem como funçao relatar o caso de uma paciente com doença autoinflamatória, e a partir dele estabelecer um fluxograma diagnóstico modelo para auxílio no diagnóstico destas doenças raras.
RELATO DE CASO
Paciente NA, sexo feminino, 42 anos, solteira e previamente hipotireoidea, em uso de levotiroxina 75 mcg por dia, e dislipidêmica, usando rosuvastatina 10 mg. Aos 41 anos começou a apresentar alteraçoes do hábito intestinal e dor abdominal em cólica. O quadro teve piora com o surgimento de febre (temperatura máxima 38,2 °C) quando foi internada e tratada com ceftriaxona por sete dias e piroxicam para analgesia, apresentando melhora sintomática em menos de uma semana e recebendo alta sem etiologia definida. Nesta ocasiao, os exames laboratoriais da paciente mostravam leucocitose com neutrofilia, além de elevaçao das provas inflamatórias de fase aguda. Dois meses após, teve novo episódio de alteraçao do hábito intestinal com febre e agora também referia mialgia em membro inferior direito, que evoluiu com lesao cutânea e edema neste membro. O quadro foi autolimitado, nao sendo utilizado antibióticos ou anti-inflamatórios.
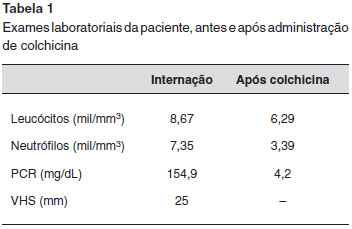
Quando a paciente procurou nosso serviço, ainda apresentava constante alteraçoes do hábito intestinal, odinofagia, cefaleia, lesoes eritematosas em face, mialgia em coxas e pernas e poliartralgia em ombros, punhos e tornozelos. A paciente negava perda de peso e se queixava de esquecimento e estresse emocional frequentes. Nao referia antecedente familiar de doenças autoimunes e neoplasias. O pai havia falecido por infarto agudo do miocárdio e a mae estava viva, com problemas cardíacos e vasculares. A paciente foi novamente internada para investigaçao. Neste momento a paciente apresentava leucocitose e neutrofilia, além de elevaçao de provas inflamatórias de fase aguda. Para que causas infecciosas, inflamatórias ou neoplásicas fossem excuídas, foi realizado PET-Scan (tomografia por emissao de pósitrons) de corpo inteiro (Figura 1). A partir desta imagem foi realizado colonoscopia para se descartar doença inflamatória intestinal. Com a ausência de alteraçoes na colonoscopia e por nao haver causa infecciosa ou neoplásica que justificassem o quadro da paciente, foi aventada a hipótese de doença autoinflamatória sistêmica, provavelmente febre familiar do Mediterrâneo e introduzido empiricamente colchicina na dose de 1,5 mg/dia. Após dois dias de tratamento, houve normalizaçao do quadro clínico e das provas inflamatórias (Tabela 1).
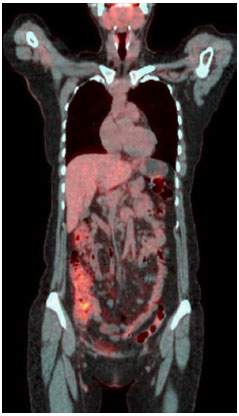
Figura 1 PET-Scan de corpo inteiro evidenciando captaçao anormal na regiao colônica. Nesta imagem observa-se ausência de captaçao no cólon ascendente e descendente, partes retroperitoneais do cólon
DISCUSSAO
As doenças autoinflamatórias estao tendo maior reconhecimento atualmente pela melhora na capacidade diagnóstica, principalmente pela evoluçao da imunologia e da biologia molecular11. O início dos sintomas e o diagnóstico em geral ocorrem na faixa pediátrica, mas podem iniciar também na fase adulta4.
Classicamente, algumas manifestaçoes clínicas sao bem relacionadas às doenças autoinflamatórias, sendo elas11: febre recorrente; derrames cavitários estéreis (pleurite, pericardite e/ou peritonite); diarreia crônica sem etiologia definida (exclusao de doenças inflamatórias intestinais clássicas); doença inflamatória do sistema nervoso central sem etiologia definida (exclusao de doenças autoimunes conhecidas e doenças infecciosas crônicas); osteomielite crônica recorrente nao infecciosa (exclusao de causas neoplásicas e infecciosas); urticária crônica ou dermatoses neutrofílicas; e manifestaçoes inflamatórias em pacientes com imunodeficiências primárias.
Para a definiçao desta síndrome clínica, a primeira coisa a fazer é a definiçao clínica. Neste caso, a paciente apresentava-se clinicamente com: febre, dor abdominal recorrente (peritonite), diarreia, artralgia e rash cutâneo similar à erisipela. Partindo destas manifestaçoes, se nao pensarmos inicialmente em doença autoinflamatória, o quadro é inespecífico e a primeira coisa a fazer é excluir ou confirmar outros diagnósticos. Para isto, faz-se necessário comprovaçao de que a síndrome em investigaçao cursa com "surtos estéreis". Assim, realizamos a investigaçao diagnóstica para afastar os eventuais diagnósticos diferenciais e comprovar de fato que a paciente tinha aumento de provas inflamatórias, de forma intermitente. Neste caso a paciente foi internada e excluída doenças infecciosas (surtos curtos sem evidência clínica de infecçao), doenças neoplásicas (análise de PET-Scan de corpo inteiro excluiu hipercaptaçao de locais suspeitos) e autoimunidades, pela ausência de autoanticorpos e ausência de quadro clínico compatível.
A demonstraçao de atividade inflamatória em surto é essencial. Ela pode ser sistêmica ou local. Quando em doenças sistêmicas, pode ser feita através de dosagens séricas de proteína C-reativa, velocidade de hemossendimentaçao, ferritina sérica, dosagem sérica de substância amiloide e, até mesmo, com dosagem de pool de citocinas12. A atividade inflamatória local pode ser aferida em cada sistema13-16, podendo-se demonstrar atividade inflamatória no liquor (celularidade aumentada - neutrofílica ou linfocítica - com proteinorraquia), dosagem de calprotectina fecal (evidência laboratorial de atividade inflamatória neutrofílica), dosagem de proteínas em derrames cavitários e, por fim, através da biopsia tecidual com demonstraçao de que a manifestaçao inflamatória é decorrente de células da imunidade inata. Eventualmente, a biopsia tecidual é necessária e espera-se encontrar infiltrados predominante neutrofílico com ou sem vasculite15. Neste caso, partindo deste raciocínio, a paciente apresentava a síndrome clínica de febre recorrente, pois conseguimos comprovar atividade inflamatória em surtos (PCR e VHS elevados na crise e normais nos períodos intercrises); ausência de doença autoimune clássica (ausência de manifestaçao clínica típica e autoanticorpos negativos); o PET-Scan mostrando captaçao peritoneal difusa, sugestivo de peritonite, foi de extrema utilidade para afastar hipóteses neoplásicas e infecciosas17,18, associado à colonoscopia com biopsias seriadas sem alteraçoes inflamatórias no anátomo-patológico, excluindo doença inflamatória intestinal19.
Outra ferramenta diagnóstica é a prova terapêutica. Para isto, medicamentos bloqueadores inespecíficos de citocinas sao utilizados. Em manifestaçoes graves ou com perda de funçao de órgaos vitais (coraçao, olho, pulmao, rim e SNC) administramos glicocorticoides em doses elevadas, que variam de 1 mg/kg/dia (dose imunossupressora) até doses de pulsoterapia (1 g/dia por 3 a 5 dias) até resoluçao do quadro. Em casos mais leves, utilizamos glicocorticoides em doses menores, 0,5 mg/kg/dia (dose anti-inflamatória), ou utilizamos anti-inflamatórios que atuem em células da imunidade inata, principalmente neutrófilos. Os principais anti-inflamatórios nao glicocorticoides disponíveis sao a colchicina (dose mínima de 1 mg/dia) e a indometacina (100 mg/dia). Outro fato é que estes pacientes nao apresentam resposta satisfatória com bloqueadores de células T e B (cloroquina, metotrexato, azatioprina ou ciclofosfamida), pois, por definiçao, estas doenças nao apresentam participaçao de imunidade adaptativa e nao têm linfócito T autorreativos20. Neste caso a paciente nao apresentava manifestaçao grave e apresentou resposta satisfatória e sustentada com bloqueadores da migraçao neutrofílica, a colchicina, que foi prescrita em dose adequada.
A grande dúvida clínica é quando indicar e como analisar o teste genético. O presente caso é bastante ilustrativo, pois o diagnóstico das doenças autoinflamatórias é clínico e o teste genético só deve ser utilizado como ferramenta adicional quando houver dúvida em relaçao ao diagnóstico ou por fins científicos21. Uma parcela considerável da populaçao ainda nao possui acesso a testes de análise genética, os quais têm ajudado muito o diagnóstico correto destas doenças, mas quando empregados de forma errada podem trazer consequências graves.
A Figura 2 resume o manejo diagnóstico das doenças autoinflamatórias, modelo que foi utilizado no caso apresentado e pode guiar os médicos que suspeitam dessas doenças. Inicialmente, deve-se caracterizar a síndrome clínica do paciente, excluindo causas infecciosas, neoplásicas, imunodeficiências primárias e doenças autoimunes clássicas. Exames laboratoriais que evidenciem aumento de atividade inflamatória, principalmente na crise, como PCR, VHS, ferritina e substância amiloide, sao muito importantes, além da confirmaçao histopatológica de infiltraçao inflamatória neutrofílica em lesoes de pele, osso e sistema nervoso central.
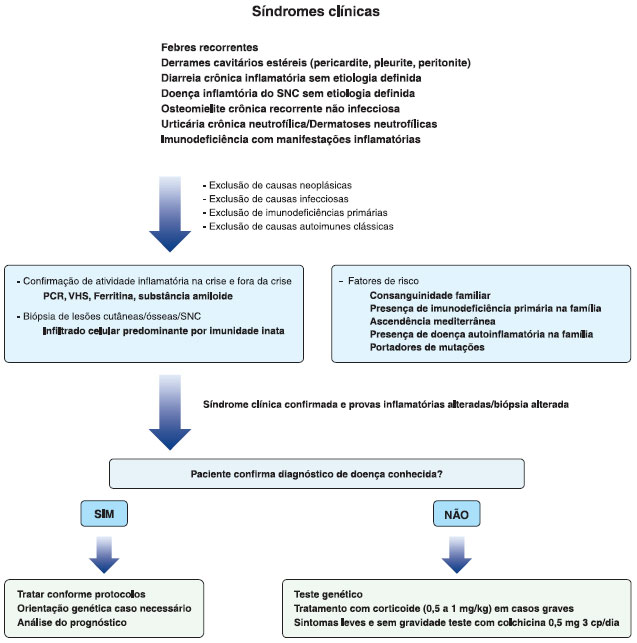
Figura 2 Manejo diagnóstico das doenças autoinflamatórias
Fatores de risco devem ser extensamente investigados, como consanguinidade, presença de imunodeficiências primárias na família, ascendência Mediterrânea/Leste Europeu e presença de doenças autoinflamatórias ou de portadores de mutaçoes na família. Com todos esses dados, em geral, é possível suspeitar-se de um diagnóstico e iniciar o tratamento conforme o protocolo da doença, fazendo orientaçao genética caso necessário. Em casos de dúvida diagnóstica, o teste genético é essencial, podendo-se utilizar glicocorticoide em doses imunossupressoras para situaçoes clínicas graves.
CONCLUSAO
As doenças autoinflamatórias sao doenças raras que precisam ser consideradas em pacientes com quadro clínico intermitente e autolimitado, com melhora importante da sintomatologia com uso de colchicina, mesmo fora da faixa etária usual - pediátrica - pela manifestaçao incompleta da doença e falha de diagnóstico anterior. O uso de PET-Scan e exames laboratoriais que evidenciem aumento de provas de atividade inflamatória sao úteis no diagnóstico. Os testes genéticos terao maior importância na confirmaçao do diagnóstico.
REFERENCIAS
1. Doria A, Dayer JM, Punzi L. Autoinflammatory diseases: How to put the fire inside the body out? Autoimmun Rev. 2012;12(1):1-4.
2. Galeazzi M, Gasbarrini G, Ghirardello A, Grandemange S, Hoffman HM, Manna R, et al. Autoinflammatory syndromes. Clin Exp Rheumatol [Internet]. 2006;24(1 Suppl 40):S79-85. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16466630 .
3. Aksentijevich I, Centola M, Deng Z, Sood R, Balow JE, Wood G, et al. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90(4):797-807.
4. Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. Am J Med [Internet]. 1967;43(2):227-53. Available from: http://www.sciencedirect.com/science/article/pii/0002934367901672 .
5. Fujikura K. Global epidemiology of Familial Mediterranean fever mutations using population exome sequences. Mol Genet Genomic Med [Internet]. 2015;3(4):272-82. Available from: http://doi.wiley.com/10.1002/mgg3.140 .
6. Berkun Y, Ben-Chetrit E. Pyrin and cryopyrin - similar domain sequence but opposite inflammatory consequence. Clin Exp Rheumatol [Internet]. Jan [cited 2016 Feb 14];25(4 Suppl 45):S6-8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17949543 .
7. Padeh S, Berkun Y. Auto-inflammatory Fever Syndromes. Rheum Dis Clin North Am. 2007;33(3):585-623.
8. Balbo BEP, Silva AA, Amaral AG, Malheiros DMAC, Onuchic LF, Barros RT. Abdominal pain, arthritis, and nephrotic syndrome in a Syrian patient. Clinics (Sao Paulo). 2012;67(6):685-8.
9. Cazeneuve C, Sarkisian T, Pêcheux C, Dervichian M, Nédelec B, Reinert P, et al. MEFV-Gene analysis in armenian patients with Familial Mediterranean fever: diagnostic value and unfavorable renal prognosis of the M694V homozygous genotype-genetic and therapeutic implications. Am J Hum Genet [Internet]. 1999 Jul [cited 2016 Feb 14];65(1):88-97. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1378078&tool=pmcentrez&rendertype=abstract .
10. Jesus AA, Oliveira JB, Hilário MOE, Terreri MTR, Fujihira E, Watase M, et al. Pediatric hereditary autoinflammatory syndromes. J Pediatr (Rio J). 2015;86(5):353-66.
11. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next-of-kin. Nat Rev Rheumatol [Internet]. Nature Publishing Group; 2014;10(3):135-47. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24247370 .
12. Berkun Y, Eisenstein EM. Diagnostic criteria of familial Mediterranean fever. Autoimmun Rev [Internet]. Elsevier B.V.; 2014;13(4-5):388-90. Available from: http://dx.doi.org/10.1016/j.autrev.2014.01.045 .
13. Cantarini L, Lopalco G, Selmi C, Napodano S, De Rosa G, Caso F, et al. Autoimmunity and autoinflammation as the yin and yang of idiopathic recurrent acute pericarditis. Autoimmun Rev [Internet]. Elsevier B.V.; 2015;14(2):90-7. Available from: http://dx.doi.org/10.1016/j.autrev.2014.10.005 .
14. Dal Pont E, D'Incà R, Caruso A, Sturniolo G-C. Non-invasive investigation in patients with inflammatory joint disease. World J Gastroenterol [Internet]. 2009;15(20):2463. Available from: http://www.wjgnet.com/1007-9327/15/2463.asp .
15. Braun-Falco M, Ruzicka T. Hautbeteiligung bei autoinflammatorischen syndromen. JDDG - J Ger Soc Dermatology. 2011;9(3):232-46.
16. Cantarini L, Rigante D, Brizi MG, Sebastiani GD, Lucherini OM, Galeazzi M, et al. The laboratory approach in the diagnosis of systemic autoinflammatory diseases. Reumatismo. 2011;63(2):101-10.
17. Chrapko BE, Chrapko M, Nocu A, Stefaniak B, Zubilewicz T, Drop A. Role of 18F-FDG PET/CT in the diagnosis of inflammatory and infectious vascular disease. Nucl Med Rev [Internet]. 2016;19(1):28-36. Available from: https://journals.viamedica.pl/nuclear_medicine_review/article/view/42253 .
18. Tokmak H, Ergonul O, Demirkol O, Cetiner M, Ferhanoglu B. Diagnostic contribution of 18F-FDG-PET/CT in fever of unknown origin. Int J Infect Dis [Internet]. International Society for Infectious Diseases; 2014;19(1):53-8. Available from: http://dx.doi.org/10.1016/j.ijid.2013.10.009
19. Hommes DW, Van Deventer SJH. Endoscopy in inflammatory bowel diseases. Gastroenterology. 2004;126(6):1561-73.
20. Wu B, Xu T, Li Y, Yin X. Interventions for reducing inflammation in familial Mediterranean fever. Cochrane database Syst Rev [Internet]. 2015 Jan [cited 2016 Feb 23];3:CD010893. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25791871 .
21. Giancane G, Ter Haar NM, Wulffraat N, Vastert SJ, Barron K, Hentgen V, et al. Evidence-based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis. 2015 Apr;74(4):635-41.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888