Número Atual: Outubro-Dezembro 2024 - Volume 8 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo Original
Nova proposta para diagnóstico do angioedema hereditário com papel filtro
New diagnostic approach to hereditary angioedema using filter paper
Maine Luellah Demaret Bardou1; Rosemeire Navickas Constantino-Silva1; Maria Luiza Oliva-Alonso2; Ana Júlia Ribeiro Teixeira3; Pedro Giavina-Bianchi3; Eli Mansour4; Solange Oliveira Rodrigues Valle2; Anete Sevciovic Grumach1
1. Centro Universitário FMABC, Imunologia Clínica - Santo André, SP, Brasil
2. Hospital Universitário Clementino Fraga Filho, Universidade Federal do Rio de Janeiro, Serviço de Imunologia, Departamento de Clínica Médica - Rio de Janeiro, RJ, Brasil
3. Universidade de São Paulo, Divisão de Imunologia Clínica e Alergia - São Paulo, SP, Brasil
4. Universidade Estadual de Campinas, Imunologia Clínica e Alergia, Departamento de Clínica Médica - Campinas, SP, Brasil
Endereço para correspondência:
Maine Luellah Demaret Bardou
E-mail: maine_bardou@hotmail.com
Submetido em: 30/05/2024
Aceito em: 04/08/2024.
RESUMO
INTRODUÇÃO: O angioedema hereditário (AEH) é raro, caracterizado por edema em subcutâneo, trato gastrointestinal e vias aéreas superiores. A deficiência do inibidor de C1 decorre da mutação em SERPING1 (AEH-C1-INH) que resulta em acúmulo de bradicinina e maior permeabilidade endotelial. O AEH também pode apresentar-se com C1-INH normal. A avaliação funcional do C1-INH permite detectar as duas formas de AEH-C1-INH. O objetivo do estudo foi avaliar a triagem de pacientes usando amostras de gota de sangue seca (DBS - Dried blood spot) em papel filtro comparada à técnica atual por ensaio cromogênico.
MÉTODOS: Estudo multicêntrico, prospectivo, em pacientes com AEH subdivididos em: G1 - AEH-C1-INH (n = 53; tipos 1 = 48 e 2 = 5); G2-AEH com mutação de FXII (AEH-FXII) (n = 30) e G3-AEH sem mutação conhecida (AEH-UNK) (n = 10) e G4 - Controle (n = 10). Realizadas dosagens de C4 e C1-INH (imunodifusão radial); C1-INH funcional (ensaio cromogênico e DBS). A avaliação estatística utilizou o programa GraphPad Prism. O protocolo foi aprovado pelo Comitê de Ética (CAAE:41812720010010082).
RESULTADOS: Valores de C4 e C1-INHq resultaram em mediana de 9,14 e 8,9 mg/dL para G1; G2 de 33,2 e 34,2 mg/dL; G3 de 35,3 e 29,4 mg/dL e G4 32,2 mg/dL e 35,3 mg/dL respectivamente. Valores de fC1-INH pelo ensaio cromogênico e por DBS resultaram em mediana de 35% e 0% em G1; G2 de 120% e 81%; G3 de 120% e 91%; e em G4 de 32,2% e 35,3%, respectivamente. G1 com 42/53 amostras (79,2%) e 53/53 (100%) com fC1-INH reduzida respectivamente. Em G2 30/30 (100%) e 28/30 (93,7%) com fC1-INH acima de 50% respectivamente. Para G3 e para G4 a fC1-INH estava normal em ambas as técnicas.
CONCLUSÃO: O fC1-INH pela técnica em DBS permite identificar os pacientes com AEH-C1-INH com maior precisão que o ensaio cromogênico. Trata-se de coleta simples, de fácil transporte, facilitando o diagnóstico de AEH-C1-INH.
Descritores: Angioedema hereditário, inibidor de C1, fator XII, C1-INH, Complemento C4, biomarcador, diagnóstico.
Introdução
Angioedema hereditário (AEH) é um termo usado para descrever episódios isolados e/ou recorrentes de angioedema em tecidos subcutâneo e submucoso, frequentemente envolvendo face, extremidades, trato gastrointestinal, genitália, vias aéreas superiores e que não apresenta associação com lesões de pele do tipo urticariformes. Frequentemente acomete muitas pessoas de uma mesma família por tratar-se de uma herança autossômica dominante com 75% com história familiar e 25% com mutação de novo1. Existem diferentes tipos de AEH, o subtipo mais frequente é o AEH com deficiência do inibidor C1 (C1-INH) (AEH-C1-INH), causado por mais de 800 mutações que afetam o gene SERPING12,3.
A maioria das mutações em SERPING1 leva a uma deficiência quantitativa na síntese e secreção de C1-INH (AEH-C1-INH tipo 1), e o diagnóstico é feito por baixos níveis plasmáticos quantitativos de C1-INH (C1-INHq). Em aproximadamente 15% dos casos de AEH-C1-INH, a mutação resulta em uma proteína anômala, detectada pela avaliação funcional de C1-INH (fC1-INH), caracterizando o AEH-C1-INH tipo 24. Nestes pacientes, os níveis quantitativos de C1-INH são normais ou mesmo elevados, enquanto os níveis de C4 e a atividade funcional de C1-INH são baixos1,4,5.
O C1-INH é inibidor de protease que inibe as vias do complemento, fibrinolítica, coagulação e cinina-calicreína; sua deficiência resulta em uma liberação descontrolada de bradicinina (BK). A BK liga-se ao receptor B2 nas células endoteliais levando à liberação do oxido nítrico e vasodilatação6,7. No entanto, existem pacientes com valores normais do C1-INH, tanto em valor como em função (AEH-nC1-INH), tendo sido associados até o momento a oito diferentes mutações. As mutações gênicas identificadas afetam o gene do fator de coagulação XII (AEH-FXII)8, plasminogênio (AEH-PLG)9, Angiopoetina-1 (AEH-ANGPT1)10, Cininogênio 1 (AEH-KNG1)11, mioferlina (AEH-MYOF)12, heparansulfato-glucosamina 3-O-sulfotransferase 6 (HS3ST6)13, carboxipeptidase N subunidade 1 (CPN1)14 e disabled homolog 2-interacting protein (DAB2IP)15,16.
Na atualidade, apesar de todas as ferramentas para o diagnóstico, a dificuldade reside no acesso aos exames, na coleta e na manipulação adequada das amostras, tendo em vista que as proteínas do sistema complemento são termolábeis, isto é, devem ser coletadas rapidamente e alíquotadas a -80 ºC para posterior avaliação laboratorial1,5,17,18. Além disso, atualmente, no Brasil há um número limitado de laboratórios clínicos que realizam a dosagem quantitativa e funcional do C1-INH19,20. Portanto, o envio das amostras de locais distantes gera um custo muito elevado, pois o transporte deve ser feito em gelo seco e em curto prazo. Assim, a disponibilização de métodos que permitam o envio de amostras sem prejuízo da avaliação funcional do C1-INH seria uma ferramenta essencial para o aprimoramento e para a equidade de acesso ao diagnóstico de AEH-C1-INH. Considerando-se o que foi apresentado, o presente estudo teve como objetivo avaliar a fC1-INH por meio de amostras em papel de filtro (DBS) para o diagnóstico de AEH-C1-INH comparado ao ensaio cromogênico.
Métodos
Trata-se de estudo multicêntrico e prospectivo, incluindo pacientes maiores de 1 ano com diagnóstico confirmado de angioedema hereditário. Para tanto, considerou-se a apresentação clínica, a história familiar positiva e os exames bioquímicos confirmatórios para pacientes com deficiência de C1-INH e/ou exames genéticos para o AEH com inibidor de C1 normal. Após a triagem inicial, 93 pacientes foram divididos em quatro grupos: (G1) AEH com deficiência de inibidor de C1 (AEH-C1-INH) tanto tipo 1 (n = 48) e quanto tipo 2 (n = 5); (G2) AEH com inibidor de C1 normal e identificação de mutação em FXII (AEH-FXII) (n = 30). (G3) AEH com inibidor de C1 normal e sem mutação conhecida (AEH-UNK) (n = 10), pois tiveram seu exoma sequenciado e não foram encontradas as mutações atuais descritas na literatura; e finalmente (G4), controles n = 10.
Foram excluídos os pacientes com incapacidade de fornecer consentimento informado ou com diagnóstico de angioedema adquirido e/ou com doenças que possam resultar em distúrbios do sistema complemento como nefropatias e hepatopatias.
As amostras foram separadas por centrifugação do sangue recém-coletado a 3000 rpm por 10 minutos a 4 ºC. Estas foram imediatamente aliquotadas e armazenadas a -80 ºC para realização da dosagem de C1-INHq, de C4 e da fC1-INH pelo método cromogênico. Foram realizadas dosagens de C4 e C1-INHq por imunodifusão radial; avaliação fC1-INH por ensaio cromogênico e por gota de sangue seca (DBS).
A avaliação da atividade funcional de C1-INH pelo método cromogênico foi realizado com o kit comercial Technochrome C1-INH®. Por esta técnica, há incorporação do substrato C1s sintético para medir a atividade inibitória da proteína C1-INH na amostra de plasma. A fC1-INH pelo método cromogênico é medida pela reação entre C1s e seu substrato artificial, Z-Lys-SBzl·HCl, para produzir cbz-Lys e tiometil benzeno. O tiometil benzeno é medido por um ensaio cromogênico, após derivatização com 5,5'-ditiobis-(ácido 2-nitrobenzoico). Uma menor intensidade de cor indica maior atividade do inibidor de C1, pois significa que há menos produção de tiometil benzeno, que é um produto da reação envolvendo o C1s e o substrato, portanto, a intensificação da cor no teste cromogênico sugere uma redução na função do inibidor de C1.
O sangue também foi coletado em tubos Vacutainer EDTA. Após a coleta, os tubos foram invertidos para ressuspender as células sanguíneas e alíquotas de 60 μL colocadas no ponto do papel de filtro, e este foi seco por pelo menos 3 horas em temperatura ambiente e armazenadas a -20 ºC por até 180 dias e enviadas para PerkinElmer Genetics para realização da fC1-INH por meio do DBS.
A coleta em papel filtro (DBS: dried blood spot) foi realizada em material padronizado fornecido por Perkin Elmer Genetics, onde se utilizou a Cromatografia Líquida acoplada à Espectrometria de Massas (LC-MS/MS) que é capaz de medir fC1-INH. Neste ensaio primeiro é realizada a extração de C1-INH dos cartões DBS; depois liga-se o C1-INH do cartão com excesso de C1s. Após é realizada uma reação de C1s que não se ligou nas etapas anteriores com seu substrato. Isto gera a formação de cbz-Lys e tiometil benzeno. O produto cbz-Lys é analisado diretamente por LC MS/MS. Assim, este método mede a atividade inibitória de C1 esterase (C1s) pelo C1-INH presente nas amostras21.
Com relação aos valores de normalidade foram considerados para C4 16,7 a 38,5 mg/dL para mulheres e de 16,2 a 44,5 mg/dL para homens; para valores de C1-INHq considerado normais entre 19,5 e 34,5 mg/dL. Para fC1-INH, os valores de normalidade segundo o kit Technochrome era acima de 69%, e para o DBS, acima de 62,8%, segundo PerkinElmer Genetics. Porém, para o diagnóstico consideram-se valores acima de 50% de acordo com a literatura3,17,22.
O estudo foi aprovado pelo Comitê de Ética em Pesquisa do Centro Universitário FMABC (CAAE:41812720010010082). Os pacientes e voluntários assinaram aos termos de Consentimento Livre e Esclarecido (TCLE) e/ou Termo de Assentimento Livre Esclarecido (TALE) antes da inclusão no estudo.
Resultados
Os pacientes avaliados eram predominantemente do sexo feminino para todos os grupos. No G1, a idade média foi de 40 (±17,7) anos para AEH-C1-INH tipo 1, e de 27 (±13,4) anos com AEH-C1-INH tipo 2. Para o G2, a ideia média foi de 38 (±18,6) anos, e em G3 foi 40 (±14,4) anos.
A maioria dos pacientes em todos os grupos era sintomática ao diagnóstico, com idade média de aparecimento dos sintomas variando de 10 a 26 anos (Tabela 1).
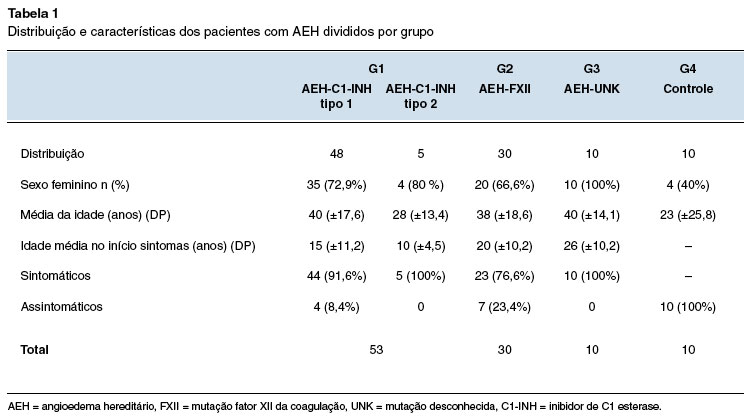
As medianas dos níveis séricos de C4 e C1-INHq resultaram para G1 em 9,14 e 8,9 mg/dL; para G2 em 33,2 e 34,2 mg/DL; e para G3 de 35,3 e 29,4 mg/dL, respectivamente (Tabela 2).
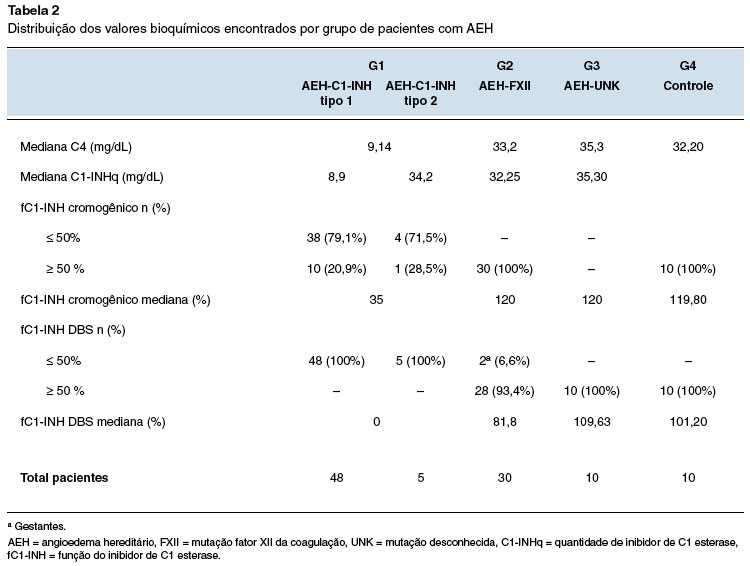
Dois pacientes com AEH-C1-INH tipo 1 apresentaram valores de C4 acima do normal (1 criança de 4 anos e 1 adulto de 27 anos). Para AEH-C1-INH tipo 2 todos apresentaram valores reduzidos de C4, e em G2, G3 e G4 os valores estavam normais (Figura 1).
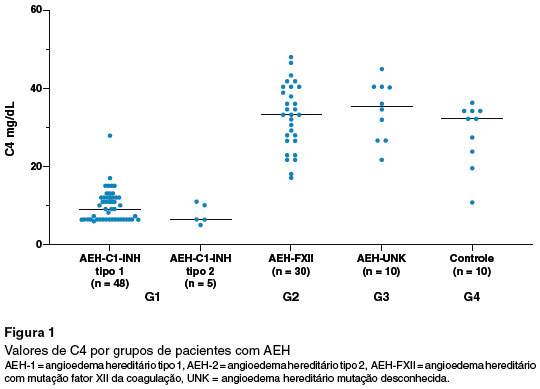
Os valores de C1-INHq encontraram-se reduzidos em 100% dos pacientes AEH-C1-INH tipo 1 e elevado nos 5 pacientes com AEH-C1-INH tipo 2, assim como em G2, G3 e G4 (Figura 2).
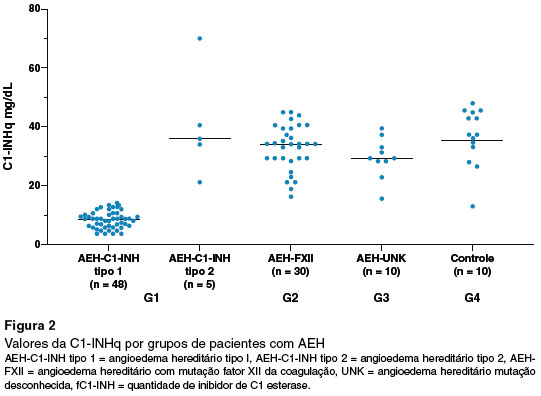
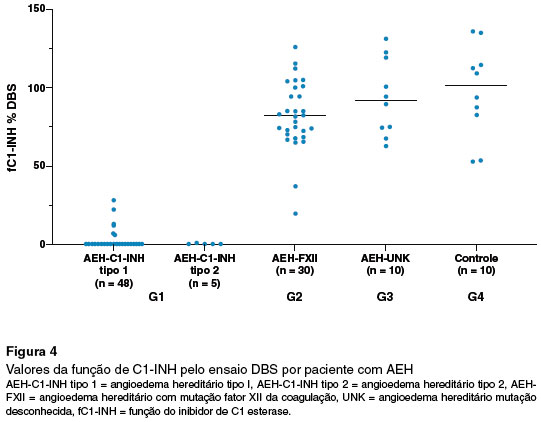
Para fC1-INH pelo ensaio cromogênico, as medianas foram de 35% para G1; de 120% para G2 e G3. Para o fC1-INH por DBS, a mediana foi de 0% para G1; de 81% para G2 e de 91% para G3 (Tabela 2).
Na avaliação individualizada da fC1-INH pelo ensaio cromogênico, G1 apresentou valores abaixo de 50% em 42 dos 53 pacientes (79,2%), sendo 10 pacientes com AEH-C1-INH tipo 1 e 4 pacientes AEH-C1-INH tipo 2 com valores acima de 50%. Para G2, G3 e G4, o fC1-INH encontrava-se acima de 50% (Tabela 2 e Figura 3).
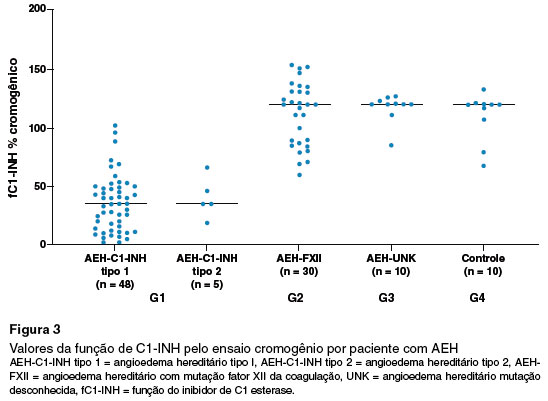
Com relação aos valores da fC1-INH pelo método com DBS, no G1 100% dos pacientes apresentaram valores abaixo de 50%. No G2 duas gestantes apresentaram resultados reduzidos da função contra 28 pacientes com resultados normais. Para G3 e G4 todos os valores estavam acima de 50 % (Tabela 2 e Figura 4).
Discussão
O angioedema hereditário é uma doença potencialmente fatal quando não diagnosticada e tratada adequadamente, com taxa de mortalidade de 25 e 40%, como decorrência do edema de glote e asfixia (23, 24) Um estudo de mortalidade por AEH verificou um taxa de mortalidade em torno de 17,5%, com apenas 13% dos pacientes com diagnóstico de AEH antes da morte25. Isto é reflexo de uma miríade de fatores como a capacitação médica em reconhecer a doença, o número reduzido de especialistas, a dificuldade de acesso ao tratamento e ao acesso aos exames diagnósticos20,26. Por exemplo, no Brasil, no Sistema Único de Saúde apenas dois laboratórios são capacitados a realizar a totalidade dos exames de diagnóstico27 que compreendem a dosagem de C4, C1-INHq e a fC1-INH. Além disso, é de suma importância que na avaliação desses componentes do complemento, as amostras de sangue sejam processadas o mais rápido possível no soro e/ou no plasma EDTA. Se a análise não for realizada imediatamente, as amostras precisam ser congeladas a -80 ºC até serem analisadas ou enviadas em gelo seco para estes laboratórios especializados. Outro ponto é que repetir o congelamento e o descongelamento pode levar a ativação in vitro do complemento18.
Atualmente, para o diagnóstico de pacientes com suspeita de AEH pode-se iniciar a investigação solicitando C4 como rastreio, disponível em vários laboratórios. Entretanto, esta estratégia revelou-se de baixa especificidade, pois o C4 ainda pode ser normal em 27% dos indivíduos que apresentam AEH-C1-INH estabelecido28,29. Outro dado relevante é a solicitação isolada do C1-INHq; estará reduzido apenas nos pacientes com AEH-C1-INH tipo 1. Desta maneira, mensurar a fC1-INH torna-se importante para fins de diagnóstico devido ao seu potencial para diagnosticar ambos os tipos de AEH-C1-INH tipo 1 e 2.
Diante desse difícil cenário, ficou evidente neste estudo que condições como recebimento de amostras de locais distantes, a forma da coleta e as condições climáticas, podem influenciar no diagnóstico de pacientes com AEH. Onze pacientes do G1 (AEH-C1-INH) em nosso estudo apresentaram valores normais de fC1-INH pelo ensaio cromogênico. Com a utilização do DBS (papel filtro), todos os 53 pacientes apresentaram C1-INHf < 50% como esperado para o G1 (AEH-C1-INH), mesmo com o envio das amostras para outro país e com tempo longo após a coleta. Estas foram mantidas a -20 ºC (freezer comum) e processadas em até 180 dias (6 meses), como visto sem prejuízo para os resultados.
Do ponto de vista metodológico, o ensaio cromogênico é baseado em duas etapas de reações, enquanto o ensaio LC-MS/MS mede o produto direto da reação C1s, portanto, a variação do ensaio pode ser menor21.
Dois pacientes com AEH-C1-INH tipo 1 apresentaram valores de C4 acima do normal, isto é, 4% dos pacientes com AEH-C1-INH tipo 1 poderiam permanecer sem diagnóstico se apenas esta triagem fosse utilizada. Vale ressaltar que a padronização da amostra em DBS demonstrou que o fC1-INH no sangue total permaneceu estável por 7 dias a 4 ºC, por até 3 dias quando armazenado a 45 ºC, e 134 dias quando armazenado em temperatura ambiente21, podendo representar uma alternativa de envio das amostras, pois exigiria um transporte com custo menor.
Vale ressaltar ainda, que neste estudo a técnica de DBS foi aplicada a pacientes com inibidor de C1 normal, pois o estudo para validação da técnica utilizou apenas pacientes com déficit em C1-INH. Observado de maneira interessante que no grupo com AEH-nC1-INH e mutação de FXII, mulheres grávidas apresentaram redução do fC1-INH por DBS (< 50%).
Conclusão
O estudo demostrou que a fC1-INH, avaliado pela técnica do DBS, é confiável para diagnosticar pacientes com angioedema hereditário com inibidor de C1 reduzido quando comparada ao teste cromogênico. Este ensaio oferece reprodutibilidade superiore para diagnóstico de pacientes com AEH-C1-INH. Trata-se de uma coleta simples, de fácil transporte e termoestáveis, oferecendo reprodutibilidade e maior precisão para o diagnóstico de AEH-C1-INH.
Referências
1. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69(5):602-16.
2. Busse PJ, Christiansen SC. Hereditary Angioedema. N Engl J Med. 2020;382(12):1136-48.
3. Maurer M, Magerl M, Betschel S, Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy. 2022 Jul;77(7):1961-90.
4. Longhurst HJ, Bork K. Hereditary angioedema: an update on causes, manifestations and treatment. Br J Hosp Med (Lond). 2019;80(7):391-8.
5. Wagenaar-Bos IG, Drouet C, Aygören-Pursun E, Bork K, Bucher C, Bygum A, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods. 2008;338(1-2):14-20.
6. López Lera A. Pathophysiology and underlying mechanisms in hereditary angioedema. Balkan Med J. 2021;38(2):82-8.
7. Sainz IM, Pixley RA, Colman RW. Fifty years of research on the plasma kallikrein-kinin system: from protein structure and function to cell biology and in-vivo pathophysiology. Thromb Haemost. 2007;98(1):77-83.
8. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343(4):1286-9.
9. Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2018;73(2):442-50.
10. Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141(3):1009-17.
11. Bork K, Wulff K, Rossmann H, Steinmüller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. 2019;74(12):2479-81.
12. Ariano A, D'Apolito M, Bova M, Bellanti F, Loffredo S, D'Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy. 2020;75(11):2989-92.
13. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. 2021;148(4):1041-8.
14. Vincent D, Parsopoulou F, Martin L, Gaboriaud C, Demongeot J, Loules G, et al. Hereditary angioedema with normal C1 inhibitor associated with carboxypeptidase N deficiency. J Allergy Clin Immunol Glob. 2024;3(2):100223.
15. D'Apolito M, Santacroce R, Vazquez DO, Cordisco G, Fantini CA, D'Andrea G, et al. DAB2IP associates with hereditary angioedema: Insights into the role of VEGF signaling in HAE pathophysiology. J Allergy Clin Immunol. 2024 Sep;154(3):698-706.
16. Germenis AE, Rijavec M, Veronez CL. Leveraging Genetics for Hereditary Angioedema: A Road Map to Precision Medicine. Clin Rev Allergy Immunol. 2021;60(3):416-28.
17. Germenis AE, Cicardi M. Driving towards Precision Medicine for angioedema without wheals. J Autoimmun. 2019;104:102312.
18. Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. 2014;61(2):110-7.
19. Veronez CL, Mendes AR, Leite CS, Gomes CP, Grumach AS, Pesquero JB, et al. The Panorama of Primary Angioedema in the Brazilian Population. J Allergy Clin Immunol Pract. 2021;9(6):2293-304.e5.
20. Serpa FS, Cruz AAS, Condino Neto A, Silva ECF, Franco JM, Mello JML, et al. O atendimento médico de pacientes com doenças imunoalérgicas no Brasil: reflexões e propostas para a melhoria - Carta de Belo Horizonte. Arq Asma Alerg Imunol. 2017;1(4):327-34.
21. Lai Y, Zhang G, Zhou Z, Inhaber N, Bernstein JA, Chockalingam PS, et al. A novel functional C1 inhibitor activity assay in dried blood spot for diagnosis of Hereditary angioedema. Clin Chim Acta. 2020;504:155-62.
22. Betschel S, Badiou J, Binkley K, Borici-Mazi R, Hébert J, Kanani A, et al. The International/Canadian Hereditary Angioedema Guideline. Allergy Asthma Clin Immunol. 2019;15:72.
23. Bork K, Siedlecki K, Bosch S, Schopf RE, Kreuz W. Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clin Proc. 2000;75(4):349-54.
24. Giavina-Bianchi P, Arruda LK, Aun MV, Campos RA, Chong-Neto HJ, Constantino-Silva RN, et al. Brazilian Guidelines for Hereditary Angioedema Management - 2017 Update Part 1: Definition, Classification and Diagnosis. Clinics (Sao Paulo). 2018;73:e310.
25. Minafra FG, Gonçalves TR, Alves TM, Pinto JA. The Mortality from Hereditary Angioedema Worldwide: a Review of the Real-World Data Literature. Clin Rev Allergy Immunol. 2022;62(1):232-9.
26. Serpa FS, Urrutia-Pereira M, Costa E, DiGesu RW, Guidacci MFRC, Cruz AS, et al. A especialidade de Alergia e Imunologia Clínica nos diferentes níveis de atenção à saúde no Brasil. Arq Asma Alerg Imunol. 2018;2(3):335-43.
27. Grupo de Estudos Brasileiro em Angioedema Hereditário, GEBRAH [Internet]. Disponível em: https://gebraeh.com.br/diagnostico-laboratorial/. Acessado em: maio/2024.
28. Tarzi MD, Hickey A, Förster T, Mohammadi M, Longhurst HJ. An evaluation of tests used for the diagnosis and monitoring of C1 inhibitor deficiency: normal serum C4 does not exclude hereditary angio-oedema. Clin Exp Immunol. 2007;149(3):513-6.
29. Charest-Morin X, Betschel S, Borici-Mazi R, Kanani A, Lacuesta G, Rivard G-É, et al. The diagnosis of hereditary angioedema with C1 inhibitor deficiency: a survey of Canadian physicians and laboratories. Allergy, Asthma & Clinical Immunology. 2018;14(1):83.
Agradecimentos: À Takeda pelo apoio financeiro e de infraestrutura, aos centros participantes e à equipe da FMABC pelo trabalho realizado.
Fonte de financiamento: Maine L. D. Bardou recebeu bolsa de iniciativa do pesquisador do LASID em 2022-2023 (Takeda Scholarships). Anete S. Grumach é bolsista de produtividade CNPq (309824/2021-4) e possui bolsa de apoio à pesquisa de iniciativa do investigador pela Shire/Takeda.
Conflito de interesses: Maine L. D. Bardou e Ana Júlia Ribeiro Teixeira declaram não possuírem conflitos de interesses. Anete S. Grumach é palestrante e/ou consultora para Takeda, CSL Behring, Pharvaris, Kalvista, Exeltis, Pint-Pharma, Biomarin, Binding site e Multicare. Possui Bolsa de Produtividade do CNPq e bolsa de apoio à pesquisa de iniciativa do investigador pela Shire/Taked. Maria Luisa O. Alonso, Pedro Giavina-Bianchi, Solange O. R. Valle e Eli Mansour receberam apoio financeiro e/ou honorários da Takeda e CSL Behring.
Trabalho vencedor do "Prêmio Antônio de Oliveira Lima" concedido no L Congresso Brasileiro de Alergia e Imunologia da ASBAI em 2023, Maceió, Alagoas, Brasil.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888