Número Atual: Abril-Junho 2019 - Volume 3 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
Deficiência de anticorpos específicos antipolissacarídeos
Specific polysaccharide antibody deficiency
Wilma Carvalho Neves Forte1; Renata Yumi Lima Konichi1; Fernanda Mezzacapa Sousa1; Tainá Mosca1; Almerinda Maria Rego2; Ekaterini Simoes Goudouris3
DOI: 10.5935/2526-5393.20190021
1. Faculdade de Ciências Médicas da Santa Casa de São Paulo, São Paulo, Disciplina de Imunologia - São Paulo, SP, Brasil
2. Universidade Federal de Pernambuco, Departamento Materno-Infantil - Recife, Pernambuco, Brasil
3. Universidade Federal do Rio de Janeiro, Departamento de Pediatria - Rio de Janeiro, RJ, Brasil
Endereço para correspondência:
Wilma Carvalho Neves Forte
E-mail: wilmanevesforte@yahoo.com.br
Submetido em: 31/01/2019
Aceito em: 25/06/2019
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
A deficiência de anticorpos específicos antipolissacarídeos é um dos erros inatos da imunidade predominantemente de anticorpos, destacando-se entre os defeitos mais frequentes. É caracterizada por uma permanência de imaturidade da resposta imunológica a antígenos polissacarídeos, estando normais linfócitos B, classes e subclasses de imunoglobulinas. O paciente apresenta maior suscetibilidade a infecções por bactérias encapsuladas, especialmente Streptococcus pneumoniae e Haemophilus influenzae. As principais manifestações clínicas são otites, sinusites, traqueobronquites e pneumonias de repetição; pode haver meningite pneumocócica e septicemia. A investigação é feita por titulação de anticorpos antipolissacarídeos antes e após a aplicação da vacina pneumocócica não conjugada. Até dois anos, há imaturidade fisiológica desse setor da imunidade, por isso, o diagnóstico não pode ser feito antes desta idade. O tratamento, além de antibiótico precoce em vigência de quadros infecciosos, inclui antibióticos profiláticos, aplicação de vacina conjugada com proteínas e/ou reposição de imunoglobulina humana endovenosa ou subcutânea. O diagnóstico e o tratamento precoce melhoram a qualidade de vida do paciente, diminuindo o risco de sequelas e até de óbito por infecção, e quando não são precoces, é possível que haja sequelas como bronquiectasias, hipoacusia ou danos neurológicos.
Descritores: Síndromes de imunodeficiência, sinais e sintomas, imunoglobulina G, vacinas pneumocócicas.
INTRODUÇÃO
Streptococcus pneumoniae e Haemophilus influenzae são os principais agentes etiológicos de pneumonias e otites. Apresentam cápsula envoltóriaformada por polissacarídeos - carboidratos contendo moléculas simples de açúcares. Para a defesa contra esses patógenos são necessários anticorpos antipolissacarídeos, contidos principalmente na subclasse IgG21,2. Tais anticorpos atuam como opsoninas, revestindo as bactérias e permitindo a fagocitose, em especial por neutrófilos.
A deficiência de anticorpos específicos antipolissacarídeos, ou simplesmente deficiência de anticorpos específicos, é uma imunodeficiência primária (IDP) (ou erro inato da imunidade - EII) com defeito predominantemente na produção de anticorpos. É caracterizada por permanência de imaturidade da resposta a antígenos polissacarídeos, estando normais os valores de linfócitos B e as concentrações séricas de classes e subclasses de IgG. Até dois anos de idade há uma imaturidade fisiológica da formação de anticorpos antipolissacarídeos. Sabe-se ainda que a maturidade em relação às subclasses de IgG é atingida por volta de quatro a seis ou sete anos de idade3,4. Por tais razões, o diagnóstico da deficiência de anticorpos específicos não pode ser feito antes de dois anos. Tendo em vista a imaturidade fisiológica3,4, estudiosos do assunto têm considerado que o diagnóstico desta deficiência pode ser feito com maior certeza após os 4 anos de idade, devendo ser considerado entre 2 a 4 anos em crianças com infecções recorrentes ou graves por bactérias encapsuladas.
A resposta deficiente de anticorpos a antígenos polissacarídeos foi descrita no final dos anos 19805, após a introdução de vacinas polissacarídeas não conjugadas para Haemophilus influenzae B46. Atualmente a deficiência específica de anticorpos destaca-se entre um dos erros inatos da imunidade mais frequentes da infância7-9.
Estimativas sugerem que seis milhões de pessoas possam estar vivendo com algum erro inato da imunidade em todo o mundo, não apenas crianças, mas também adultos10,11. Em 2014, o registro da European Society for Immunodeficiencies (ESID), estudando 19.366 crianças e adultos com IDPs, constatou que 56,66% dos pacientes apresentavam deficiências predominantemente de anticorpos; 13,91% outras IDPs bem definidas; 8,89% desordens de fagócitos; 7,47% deficiências de células T; 4,82% deficiências de complemento; 3,89% síndromes com imunodesregulação; 2,06% síndromes autoinflamatórias; 1,41% IDPs não classificadas; 1% defeitos da resposta imune12. Em 2007, o Latin American Group for Immunodeficiencies (LAGID) realizou um estudo em 3.321 pacientes com IDPs de 12 países, constatando o predomínio de deficiências de anticorpos (53,2%), das quais 4,2% apresentavam deficiência de anticorpo específico13.
No Brasil, é referido que, entre crianças com infecções respiratórias repetitivas, a prevalência de deficiência de anticorpos específicos antipolissacarídeos, varia entre 7 a 19%, representando 8,7% dos casos de defeitos de anticorpos13.
Acredita-se que na faixa etária pediátrica, aproximadamente 10% dos pacientes com infecções respiratórias recorrentes possam ter algum tipo de imunodeficiência14. A exata prevalência da deficiência de anticorpos específicos ainda não é conhecida, e talvez seja a oitava IDP mais identificada no mundo. Também não são conhecidos os mecanismos moleculares envolvidos em sua gênese7.
Uma resposta deficiente a antígenos polissacarídeos pode estar presente em outros erros inatos da imunidade, assim como em outras condições clínicas. Assim, pode estar relacionada a imunodeficiências secundárias associadas ao envelhecimento, HIV, uso de imunossupressores, doença pulmonar crônica e à asplenia congênita ou funcional, tal como ocorre na anemia falciforme.
Em estudo realizado nos EUA em 2016, avaliando o diagnóstico e o tratamento de IDPs por médicos de família e comunidade, constatou que este grupo de doenças é ainda pouco conhecido. O estudo concluiu que tais profissionais não se sentiam capacitados para identificá-las15.
É sempre importante considerar que a falta de diagnóstico, o diagnóstico tardio ou a falta de tratamento específico desta IDP pode acarretar sequelas irreversíveis pulmonares, auditivas e neurológicas, assim como acentuado aumento da mortalidade. Além do prejuízo claro aos pacientes, essas condições levam a maior gasto com atendimentos de emergência, hospitalizações e tratamento de sequelas. Tudo isso pode ser evitado com o diagnóstico e tratamento precoces desse defeito imunológico16,17.
Em muitos países, o conhecimento adequado das IDPs é escasso. As autoridades de saúde são pouco informadas e, por diversas vezes, subestimam o quadro epidemiológico destas deficiências, dificultando a atuação de comunidades médicas que atuam nesse campo. Uma maior divulgação poderia prover um atendimento mais adequado e de maior qualidade aos pacientes18.
Desse modo, continua sendo de extrema relevância o estudo e a divulgação do diagnóstico e tratamento das deficiências imunológicas. Tendo em vista a importância do diagnóstico e tratamento precoces da deficiência de anticorpos específicos antipolissacarídeos, assim como o possível impacto na saúde e em políticas públicas, nos propusemos a realizar a presente revisão.
OBJETIVO
Revisão da literatura sobre deficiência de anticorpos específicos, incluindo a resposta imunológica a antígenos polissacarídeos, assim como características clínicas, fisiopatológica, diagnóstica e tratamento deste erro inato da imunidade.
MÉTODOS
Revisão não sistemática de literatura de 2005 a 2018, com base em dados MEDLINE, LILACS, SciELO, PubMed, assim como consulta a arquivo de publicações da Associação Brasileira de Alergia e Imunologia. Foram utilizados como descritores: deficiência de anticorpos específicos; anticorpos polissacarídeos; síndromes de imunodeficiência.
RESULTADOS DA REVISÃO
Resposta imunológica a polissacarídeos de cápsulas bacterianas
Os diplococos Gram-positivos Streptococcus pneumoniae e os bacilos Gram-negativos Haemophilus influenzae apresentam cápsula envoltória polissacarídea, proporcionando maior virulência. São bactérias comensais que habitam a nasofaringe de humanos saudáveis e têm a capacidade de migrar desse nicho anatômico e causar diferentes doenças19. S. pneumoniae são os principais agentes etiológicos de pneumonias, podendo causar a doença em qualquer idade, enquanto H. influenzae causam infecções principalmente em crianças pequenas.
Conhecer os mecanismos imunológicos de resposta aos antígenos polissacarídeos é importante para que melhor se compreenda a investigação diagnóstica do tipo de deficiência imunológica da qual aqui tratamos.
Os polissacarídeos bacterianos são antígenos T independentes, o que significa que a resposta a eles se dá por meio de reconhecimento direto por imunoglobulinas presentes na circulação ou na superfície de linfócitos B H. influenzae20. A defesa imunológica a estes agentes infecciosos confere proteção e implica essencialmente na atuação de opsoninas que irão revestir a cápsula polissacarídea levando à adesão, ingestão, digestão e eliminação dos patógenos por células fagocíticas, em especial neutrófilos H. influenzae21.
Algumas características desta resposta são relevantes: não há desenvolvimento de células T de memória (o que implica memória de menor duração e ausência de resposta ampliada diante de segunda exposição) e há uma restrição de isotipos de anticorpos produzidos (basicamente IgM e IgG2)22. Há desenvolvimento de linfócitos B de memória diferentes daqueles produzidos por antígenos proteicos e cuja ativação secundária por antígenos polissacarídeos é controlada por meio de anticorpos IgG específicos23.
A ligação do polissacarídeo às proteínas do surfactante inicia a defesa contra bactérias encapsuladas, permitindo a opsonização de parte destas bactérias. Sequencialmente, receptores para lecitinas, como specific intercelular-adhesion-molecula non integrina receptor (SIGN-R1 ou CD209b), que existem em alguns macrófagos, unem-se a polissacarídeos capsulares. O complexo SIGN-R1/polissacarídeos, expresso na superfície de macrófagos, ativa diretamente o sistema complemento, mesmo na ausência de anticorpos, resultando na geração de C3b ativado24.
Segue-se uma maior ativação do complemento, por via clássica, por meio de IgM sintetizada por linfócitos B. A IgM liga-se ao primeiro componente da via clássica do complemento, o C1q, resultando na ativação de C4b e C2a da cascata do complemento, com geração de C3b. O componente C3b une-se às moléculas antigênicas polissacarídicas, revestindoas. Os receptores de complemento tipo 1 (CR1 ou CD35), expressos em fagócitos, têm alta afinidade por C3b, resultando na união de C3b a CR1. A união da bactéria com cápsula polissacarídea revestida pela opsonina C3b ao receptor CR1 do fagócito permite a opsonização ou fagocitose facilitada25.
O componente C3d, produto da clivagem de C3b, dá origem ao complexo molecular polissacarídeo/ fragmento C3d. O complexo une-se a receptores CR2 (CD21) presentes em células B, estimulando tais linfócitos a sintetizarem mais IgM. O complexo C3d/polissacarídeo é um potente imunógeno, indutor de um grande número de células secretoras de anticorpos antipolissacarídeos, mesmo em baixas concentrações de antígeno26.
Anticorpos específicos contra polissacarídeos da cápsula bacteriana exercem um papel crucial na defesa imunológica, atuando como as principais opsoninas para bactérias encapsuladas. A defesa efetiva de bactérias encapsuladas requer um processo que depende da eficiência da interação entre anticorpo anticapsular e receptor Fc da imunoglobulina. Para a síntese de anticorpos antipolissacarídeos é imprescindível a participação de células da resposta imunológica adaptativa. Antígenos timo-independentes ativam o sistema CD21, CD19, CD20, CD81 e BCR (receptor de célula B) de linfócitos B para estabelecer uma resposta produtora de IgM. Posteriormente, linfócitos B e células dendríticas apresentam antígenos polissacarídeos associados a HLA-II para o complexo receptor de T (receptor de célula T ou TCR, CD3 e CD4) de linfócitos T auxiliares tipo 1. Estes, agora ativados sintetizam interferon-gama (IFN-γ). Linfócitos Th1, na presença de IFN-γ e de diferentes moléculas de adesão, em especial CD40L em T e CD40 em B, passam a interagir com linfócitos B, iniciando-se a síntese de anticorpos sorotipo-específicos da subclasse IgG2 - os principais anticorpos antipolissacarídeos que atuam como opsoninas na defesa contra polissacarídeos de bactérias encapsuladas27.
O reconhecimento inicial do patógeno polissacarídeo por fagócitos ocorre por meio de receptores destas células: receptores de reconhecimento para padrões regulares, receptores transmembrânicos Toll-like (TLR) e receptores citosólicos nucleotide-binding oligomerization domain (NOD). A interação entre os receptores, como TLR-2, TL-4, NOD-2 e polissacarídeo determina a ativação de fator nuclear de cadeias kappa de células B (NF-κB), o qual se desloca para o núcleo. O resultado é a ativação de genes promotores de síntese de proteínas da fase aguda da inflamação, de moléculas de adesão e de citocinas pró-inflamatórias, em especial IL-1, TNF, IL-6, IFN-α e MCP-1. Tais citocinas recrutam e ativam fagócitos, células dendríticas e linfócitos, aumentando a imunidade inata e promovendo a adaptativa. Os linfócitos reguladores influenciam a magnitude da resposta a anticorpos antipolissacarídeos25.
Para analisar a resposta a este tipo de antígeno bacteriano, é relevante considerar que existe uma grande variabilidade entre os polissacarídeos das cápsulas dentro de uma mesma espécie de bactéria, e entre as diferentes espécies. Além disso, muitos polissacarídeos têm pouco poder imunogênico, pois são estruturalmente semelhantes a alguns glicolipídeos e glicoproteínas presentes no corpo humano22.
Manifestações clínicas da deficiência de anticorpos específicos antipolissacarídeos
As manifestações clínicas mais frequentes desta IDP são as infecções de repetição do trato respiratório, tal como acontece nos demais defeitos predominantemente de anticorpos: otites, sinusites, traqueobronquites e pneumonias de repetição em diferentes lobos pulmonares28-30. Os quadros infecciosos em geral são causados em geral por Streptococcus pneumoniae, mas também por Haemophilus influenzae, Moraxella catarrhalis e Staphylococcus aureus36.
Complicações como meningite, osteomielite e septicemia podem ocorrer7,31. Mais raramente, pode evoluir com doenças autoimunes32,33. O diagnóstico da deficiência de anticorpos específicos muitas vezes é feito concomitante a episódios de asma ou de sibilância pós-viral, e após procedimentos cirúrgicos de vias aéreas superiores34,35.
Os principais sorotipos determinantes de pneumonias em nosso meio são em ordem de frequência: 14, 1, 5, 6B e 337. Os sorotipos mais frequentes da meningite pneumocócica são:6, 9, 14, 18 e 23. Tratase de meningite muito grave, que leva à hipoacusia em 60% dos casos, e a óbito em 20% dos pacientes pediátricos. Outras sequelas neurológicas podem surgir após a meningite pneumocócica.
As principais manifestações clínicas da deficiência de anticorpos específicos antipolissacarídeos encontram-se entre os dez sinais de alerta para imunodeficiência primária na criança, propostos pelo Grupo Brasileiro de IDP (BRAGID), adaptados dos sinais propostos pela Fundação Jeffrey Modell e Cruz Vermelha Americana: duas ou mais pneumonias no último ano; quatro ou mais otites no último ano; um episódio de infecção grave (meningite ou septicemia). Dentre os dez sinais de alerta para imunodeficiência primária no adulto, também se encontram os que podem indicar a presença de deficiência de anticorpos específicos: uma pneumonia por ano por mais do que um ano; duas ou mais novas otites no período de um ano; duas ou mais novas sinusites no período de um ano na ausência de alergia; uso de antibiótico intravenoso de repetição para tratar infecção38.
É possível que as pneumonias de repetição tenham início em qualquer idade, inclusive em adultos, pelas mesmas razões que tal fato foi observado na imunodeficiência comum variável: mudança de ambiente físico do paciente propiciando maior contato com patógenos, como pode ocorrer após mudança de zona rural para urbana, ou ingresso em escolas maiores ou em ambientes de trabalho com maior número de pessoas39.
As pneumonias de repetição podem evoluir com sequelas pulmonares irreversíveis. Além disso, a investigação imunológica de pacientes com bronquiectasias de origem indeterminada pode levar ao diagnóstico de uma deficiência imunológica40,41. A maioria das sequelas pulmonares da deficiência de anticorpos específicos de antipolissacarídeos, como bronquiectasias, é decorrente do retardo do diagnóstico. As sequelas, de forma geral, levam a limitações do prognóstico do paciente41,42.
A deficiência na produção de anticorpos específicos para antígenos polissacarídeos pode acontecer em outras IDPs, como deficiência de IgA, síndrome de DiGeorge, candidíase mucocutânea crônica, síndrome de Wiskott-Aldrich, síndromes de HiperIgE, Imunodeficiência Comum Variável, defeitos de switch de classes ou defeito em NEMO43. Nesses casos, há outras manifestações clínicas e laboratoriais associadas. Dessa maneira, na deficiência de IgA há infecções de repetição em mucosas, em especial de vias aéreas superiores e digestório, incluindo infecções por enterovírus e giardíase. Na síndrome de DiGeorge há hipocalcemia pelo hipoparatireodismo apresentado, que leva a tremores até a convulsões no recém-nascido; depois se iniciam quadros infecciosos graves, principalmente por microrganismos oportunistas. Na candidíase mucocutânea crônica, além do quadro de pele e de mucosas por falta de defesa contra Candida albicans, com frequência o paciente apresenta endocrinopatias. Na síndrome de Wiskott-Aldrich há eczema e quadros hemorrágicos consequentes à plaquetopenia.
A diminuição de anticorpos antipolissacarídeos pode estar presente também em algumas imunodeficiências secundárias, como esplenectomia, síndrome nefrótica, enteropatias perdedoras de proteína, quilotórax, desnutrição ou imunossupressão43.
Defeito na produção de anticorpos para polissacarídeos pode ainda estar associado a quadros de alergia respiratória, particularmente rinite alérgica, ou pode mimetizá-los44. Devemos considerar esta possibilidade diante de pacientes com quadro clínico compatível com alergia respiratória, mas sem evidências laboratoriais de sensibilização alérgica ou má resposta ao tratamento específico para alergia43.
Na maioria das crianças pequenas o quadro melhora com o crescimento, constituindo-se em um maior atraso da maturação do sistema imunológico. Estima-se que o quadro esteja resolvido em até três anos em 50% dos pacientes pediátricos. A chance de resolução é menor quando o diagnóstico é feito em adolescentes ou adultos43.
Diagnóstico da deficiência de anticorpos específicos antipolissacarídeos
Os anticorpos contra a cápsula polissacarídica de S. pneumoniae e de H. influenzae representam a principal forma de proteção contra doenças invasivas por tais patógenos.
A resposta imunológica para os diferentes antígenos apresenta um processo de maturação, que ocorre dinamicamente nos primeiros anos de vida. A resposta a antígenos proteicos se estabelece bem precocemente. Entretanto, a resposta aos antígenos polissacarídeos requer um tempo mais prolongado para amadurecer, e costuma estar presente por volta dos 2 anos de idade. Além disso, linfócitos B neonatais apresentam baixas concentrações de receptores CR2, e sua aquisição ocorre com o evoluir da idade45. Portanto, a avaliação funcional de anticorpos antipolissacarídeos começa a ter valor diagnóstico a partir dos 2 anos de vida, sendo o diagnóstico de maior certeza da deficiência a partir dos 4 anos. Isto significa que é preciso ter cuidado ao fazer este diagnóstico entre os 2 e 4 anos de idade.
Além disso, a concentração de anticorpos antipolissacarídeos do tipo IgG mantém-se estável até 60 anos; entretanto, entre 61 e 90 anos há aumento significante na média da concentração sérica, quando comparada à faixa de 18 a 60 anos. A prévia vacinação ou exposição a patógenos pneumocócicos na população de idade mais avançada pode ser responsável pelos títulos mais elevados de anticorpos antipolissacarídeos. Entretanto, nesta faixa etária mais avançada, há significativa redução na concentração de IgM específica para esses antígenos, o que pode caracterizar uma menor proteção a infecções por esses microrganismos46.
Isto significa que a hipótese diagnóstica desse erro inato da imunidade pode ser considerada em qualquer faixa etária47.
A suspeita de IDP deve ter embasamento na história clínica38. Entretanto, o diagnóstico definitivo depende da realização de exames complementares. As dosagens séricas das classes de imunoglobulinas (IgM, IgG, IgA e IgE), comparadas às curvas de normalidade para cada faixa etária, permitem o diagnóstico das IDPs humorais mais frequentes. Valores normais de IgG, entretanto, não afastam deficiências de subclasse de IgG, em especial de IgG2. Da mesma forma, valores normais de IgG2 não afastam a deficiência de anticorpos específicos antipolissacarídeos48.
O diagnóstico da deficiência de anticorpos específicos antipolissacarídeos é estabelecido por meio da titulação reduzida de resposta a antígenos polissacarídeos para S. pneumoniae, estando normais a quantificação de linfócitos B, os valores de IgG e IgG2 séricas e de anticorpos contra antígenos proteicos (toxoides tetânico e diftérico, por exemplo)49. Os critérios para definir resposta adequada e inadequada a estes antígenos ainda são motivo de debate7.
Não temos disponíveis vacinas não conjugadas a proteínas para N. meningitidis ou H. influenzae, portanto não é possível usar a sorologia para esses agentes infecciosos para a pesquisa de deficiência na produção de anticorpos para polissacarídeos. A vacina pneumocócica não conjugada é, atualmente, a única forma de analisar a resposta a antígenos polissacarídeos50,51, uma vez que a vacina conjugada refletirá apenas a qualidade da resposta a antígenos proteicos52,53.
A vacina não conjugada disponível atualmente é 23-valente, e contém sorotipos que representam mais de 80% dos sorotipos responsáveis por doenças pneumocócicas invasivas no Brasil. Portal motivo, é a vacina mais indicada para o diagnóstico do erro inato da imunidade em estudo (Figura 1).
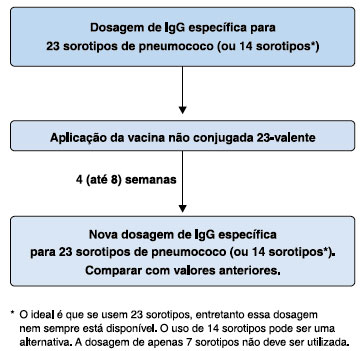
Figura 1 Algoritmo para investigação diagnóstica
Diferentemente da vacina com polissacarídeos não conjugados, na vacina conjugada específica contra bactérias encapsuladas, proteínas são quebradas em peptídeos e associadas a moléculas de superfície do complexo de histocompatibilidade classe II. Na sequência, há apresentação às células T, que estimulam a síntese de imunoglobulinas por células B. Assim, através da conjugação, o polissacarídeo pouco imunogênico adquire a característica imunogênica da proteína, sendo reconhecido pelo sistema imunológico como T-dependente, resultando de imunização em mucosas e memória imunológica54. Por tais razões, a vacina conjugada induz imunidade regular contra doenças invasivas e defesa parcial contra as não doenças invasivas, inclusive em crianças. Nesse contexto, polissacarídeos capsulares conjugados a proteínas como o toxoide tetânico, toxoide diftérico, variante mutada da toxina diftérica e vesículas de membrana externa de N. meningitidis são utilizados como antígenos vacinais contra H. influenzae tipo b, N. meningitidis e S. pneumoniae55,56.
A vacina pneumocócica conjugada 13 valente é aplicada em clínicas privadas de imunização e composta pelos sorotipos: 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F e 23F. A vacina conjugada 10-valente é utilizada no Programa Nacional de Imunizações está disponível em unidades de saúde e é composta pelos sorotipos 1,4, 5, 6B, 7F, 9V, 14, 18C, 19F, 23F conjugados à proteína de H. influenzae NT, um sorotipo conjugado ao toxoide tetânico (18C) e um conjugado ao toxoide diftérico (19F)57.
Os sorotipos existentes na vacina pneumocócica 23 valente (não conjugada) são: 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 10A, 11A, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F e 33F 58,59. Ou seja, na vacina não conjugada há 10 sorotipos não presentes na vacina conjugada 13 valente, e 13 sorotipos não presentes na vacina conjugada 10-valente (Tabela 1).

O exame para avaliação da resposta a antígenos polissacarídeos analisa a quantidade de anticorpos específicos contra os sorotipos de S. pneumoniae ausentes nas vacinas conjugadas60-62. Com a dosagem de 7 sorotipos disponível na maioria dos laboratórios de análises clínicas, apenas é possível analisar a resposta a antígenos proteicos, pois estes 7 sorotipos estão presentes em todas as vacinas conjugadas (7, 10, 13 valente). Para que se possa avaliar a resposta a antígenos polissacarídeos, é necessário utilizar a dosagem de 14 ou, idealmente, 23 sorotipos de pneumococos, com as quais é possível analisar, respectivamente, a resposta a 3 ou 10 sorotipos não presentes em vacinas conjugadas utilizadas em nosso meio. Em pacientes que não receberam qualquer vacina conjugada previamente, todos os sorotipos podem ser utilizados na interpretação (Tabela 2).

Estudos realizados nos anos 1980, por radioimunoensaio (RIE), sugeriam que valores de anticorpos específicos em torno de 2 µg/mL, induzidos por vacinação, seriam protetores contra doença pneumocócica invasiva. Métodos mais recentes (ELISA e imunoensaios múltiplos), que incorporam uma etapa para remoção de anticorpos que possam produzir reações cruzadas, permitem resultados comparáveis entre si, porém mais baixos do que os resultados obtidos por RIE63. A proteção conferida por estes anticorpos depende não só da quantidade produzida, mas também da avidez e da atividade opsonofagocítica64.
Apesar das controvérsias encontradas na literatura, tem-se admitido que uma resposta apropriada de anticorpos antipolissacarídeos, caracterizada por:
- valores iguais ou superiores a 1,3 µg/dL para cada sorotipo após vacinação não conjugada contra S. pneumoniae, em um mínimo de 50% (crianças abaixo de 6 anos), e 70% (acima de 6 anos) dos sorotipos polissacarídeos analisados;
- títulos da resposta vacinal que duplicam em um mínimo de 50% (abaixo de 6 anos) e 70% (acima de 6 anos) dos sorotipos antipolissacarídeos analisados, depois de 4 a 6 semanas de vacinação não conjugada, quando comparados aos títulos pré-vacinais do paciente7,48,65.
O mais importante é averiguar se os níveis após a imunização estão acima do valor protetor. O critério de duplicação dos valores é aplicado em caso de nível pré-vacinal acima dos níveis protetores. Em caso de níveis muito baixos, a duplicação dos valores pode ser irrelevante. Mas, em caso de níveis muito elevados na dosagem pré-vacinal, é pouco provável que estes valores aumentem muito após a imunização7. A deficiência de produção de anticorpos específicos para polissacarídeos é considerada grave quando há resposta a apenas dois ou menos sorotipos de pneumococo. É considerada moderada quando há resposta a menos de 50% dos sorotipos em menores de 6 anos, e menos de 70% dos sorotipos em maiores de 6 anos. O defeito é leve quando não há boa resposta a 50% dos sorotipos em menores de 6 anos, e a 70% dos sorotipos em maiores de 6 anos7,50.
A ausência de resposta a todos os sorotipos no exame pré-vacinal, ainda que não seja comum, não nos permite fazer qualquer diagnóstico, uma vez que a memória imunológica para antígenos polissacarídeos não costuma ser tão duradoura quanto aquela para antígenos proteicos. Há, no entanto, outro fenótipo dessa deficiência específica da produção de anticorpos que é a perda precoce da memória imunológica. Neste caso, o paciente apresenta resposta apropriada na dosagem pós-vacinal, que é perdida após 6 meses.
Importante ressaltar que as dosagens de anticorpos antes e após a vacinação devem ser feitas no mesmo laboratório, para evitar interferências referentes a diferenças de métodos utilizados. Além disso, é muito importante que a segunda dosagem seja feita entre 4 e 8 semanas após a aplicação da vacina. Intervalos maiores podem produzir resultados falsamente negativos, ou valores mais baixos.
Se a investigação não for conduzida seguindo estas orientações, nova aplicação da vacina não conjugada só é possível depois de 3 anos, pelo maior risco de efeitos adversos relevantes, particularmente locais.
Tratamento da deficiência de anticorpos específicos antipolissacarídeos
O tratamento recomendado se baseia na experiência clínica de especialistas e inclui terapia antimicrobiana, que deve ser feita logo ao início das infecções ou de maneira regular e contínua profilaticamente, uso de vacina antipneumocócica conjugada e/ou reposição de imunoglobulina. Quadros associados à de alergia respiratória devem ser adequadamente tratados, o que pode contribuir para uma frequência menor de infecções7.
Não existe um consenso sobre critérios para uso de antibioticoterapia profilática, assim como não há consenso sobre qual esquema de antibiótico deve ser usado em cada situação. A antibioticoterapia profilática pode ser realizada com azitromicina, sulfametoxazol-trimetropim, ou ainda amoxicilina, em dose plena, uso regular e contínuo, tal como em outros defeitos predominantemente de anticorpos. Alguns pacientes apresentam mais sintomas em alguns períodos do ano e podem receber antibióticos profiláticos apenas nestes períodos.
Diante da ausência de resposta à vacina não conjugada, está indicada a aplicação da vacina conjugada com o maior número de sorotipos que estiver disponível (13-valente, em nosso meio). Alguns autores indicam a aplicação de nova dose da vacina não conjugada um ano depois42.
Os pacientes devem ser reavaliados a cada seis meses sobre a continuidade da antibioticoterapia. Se não há adequado controle dos quadros infecciosos e/ou não há boa tolerância a seu uso contínuo, é preciso considerar o início da reposição de imunoglobulina humana, que pode reduzir o aparecimento de novas pneumonias, sequelas pulmonares e outras comorbidades, permitindo uma melhor qualidade de vida e até mesmo a sobrevida do paciente66-69. A necessidade dessa reposição deve ser reavaliada em 1 a 2 anos, particularmente em caso de crianças de baixa faixa etária7.
A imunoglobulina humana é atualmente usada como o principal recurso terapêutico em praticamente 75% das IDPs, nas quais há comprometimento na produção de anticorpos, promovendo a reposição de imunoglobulina da classe IgG. O intuito é manter as concentrações estáveis e adequadas de IgG no plasma e um bom controle clínico dos pacientes, com a intenção de minimizar a gravidade e a frequência das infecções, bem como evitar complicações a longo prazo7,70-72.
De acordo com "II Consenso Brasileiro sobre o uso de imunoglobulina humana", há indicação de reposição em IDPs com prejuízo evidente da produção de anticorpos da classe IgG73. A European Society for Immunodeficiencies (ESID) recomenda reposição com imunoglobulina humana em situações bem determinadas: está indicada para pacientes com concentrações séricas de IgG < 200 mg/dL, exceto para aqueles com hipogamaglobulinemia transitória da infância sem ocorrência de infecções graves; para pacientes com concentrações séricas de IgG entre 200 e 500 mg/dL a reposição é indicada quando há detecção de deficiência na produção de anticorpos ou infecções recorrentes e/ou graves; para pacientes apresentando concentrações séricas de IgG > 500 mg/dL é indicada a reposição apenas quando houver defeito comprovado na produção de anticorpos específicos, associado a infecções repetitivas e graves74,75.
A imunoglobulina humana pode ser aplicada por via endovenosa ou subcutânea. A dose inicial padrão da imunoglobulina intravenosa varia de 400 a 600 mg/ kg/dose, a cada 21 dias. As doses e o intervalo de infusão devem ser ajustados de acordo com a resposta clínica e as concentrações de IgG obtidas em cada paciente, salientando a importância da adequação do tratamento a cada indivíduo. Para doença pulmonar e/ ou sinusal crônica, há a indicação de dosagens mais altas, entre 600 e 800 mg/kg/dose (ou até 1.200 mg/ kg). A administração de imunoglobulina por via endovenosa promove a elevação de concentrações séricas em poucas horas, e é da ordem de 100 a 200 mg/dL para cada 100 mg/kg de imunoglobulina aplicada. Decresce rapidamente nos primeiros dias pela redistribuição tissular, apresentando meia vida de 21 a 28 dias. Uma vez alcançado um bom controle clínico e concentrações estáveis de IgG sérica, as aplicações intravenosas podem ser feitas em um intervalo maior, de 28 dias. Geralmente, a estabilização dos valores séricos de IgG decorre no período após 3 ou até 6 meses de tratamento7,73.
A dosagem de imunoglobulina subcutânea também é de 400 a 600 mg/kg/mês, sendo aproximadamente 100 a 150 mg/kg por semana. O intervalo entre as doses pode ser desde quinzenal até diário, aplicando-se por meio de bombas de infusão ou por push. É recomendado para o início do tratamento por via subcutânea que as dosagens sejam aplicadas em intervalos menores: 5 dias seguidos, com administração de 100 mg/kg na primeira semana, ou duas vezes por semana nas duas semanas iniciais. O aumento nas concentrações séricas de IgG é estimado em 84,4 mg/dL para cada aumento de 100 mg/kg/mês na dose de imunoglobulina subcutânea. As concentrações de IgG no sangue elevamse mais lentamente quando comparadas à infusão intravenosa, com pico entre 2 a 4 dias66,73.
Um estudo de metanálise realizado em 2016 mostrou que doses mais elevadas por via subcutânea estão relacionadas ao melhor controle clínico dos pacientes76. Apresenta como vantagem: concentrações séricas de IgG mais estáveis e obtidas mais rapidamente (entre 6 e 10 semanas de uso), menos efeitos adversos sistêmicos, apesar de mais reações nos locais das infusões77. Pode ser administrada em casa, desde que as condições de higiene permitam, não necessitando atendimento hospitalar, resultando em menor custo para o sistema de saúde; a maior parte do gasto é atribuída ao próprio medicamento73,78. A aplicação domiciliar, no entanto, ainda não está aprovada em nosso país.
Independentemente do esquema terapêutico adotado, este deve ser revisto em 1 a 2 anos, uma vez que em boa parte dos pacientes, particularmente os pediátricos, uma boa resposta aos polissacarídeos se estabelece com o passar do tempo7. Vale lembrar que nova avaliação da resposta imune apenas pode ser realizada 6 a 8 meses após a interrupção da reposição regular de imunoglobulina.
CONCLUSÕES
As infecções por S. pneumoniae e H. influenzae são muito prevalentes, particularmente em crianças de baixa faixa etária, e podem ser invasivas. A defesa contra esses agentes infecciosos é dada por anticorpos que dependem da interação entre linfócitos Th1 e B, resultando na produção de anticorpos antipolissacarídeos contidos principalmente na subclasse IgG2.
A deficiência de anticorpos específicos antipolissacarídeos é um dos erros inatos da imunidade mais frequentes. As principais manifestações clínicas são otites, sinusites, traqueobronquites e pneumonias de repetição, podendo também ocorrer meningite e septicemia. Na falta de tratamento específico, as pneumonias de repetição podem evoluir para bronquiectasias e outras sequelas pulmonares irreversíveis. As meningites pneumocócicas apresentam elevada morbimortalidade.
O diagnóstico dessa condição clínica é motivo de muita controvérsia e confusão no cotidiano, e deve ser feito por meio da avaliação funcional de anticorpos antipolissacarídeos, comparando-se a titulação de anticorpos do tipo IgG antes e após a vacinação pneumocócica não conjugada.
O tratamento é feito com antibióticos administrados precocemente no início de quadros infecciosos, uso de vacina antipneumocócica conjugada, antibioticoterapia profilática e/ou reposição de imunoglobulina humana. O objetivo do tratamento precoce é prevenir o aparecimento de novas pneumonias e de novas sequelas pulmonares, melhorando a qualidade de vida e a sobrevida do paciente.
REFERÊNCIAS
1. Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:1-26.
2. Uddin S, Borrow R, Haeney MR, Moran A, Warrington R, Balmer P, et al. Total and serotype-specific pneumococcal antibody titles in children with normal and abnormal humoral immunity. Vaccine. 2006;24(27-28):5637-44.
3. Wahn U, Moises A. Calderon, Pascal Demoly. Real-life clinical practice and management of polysensitized patients with respiratory allergies: a large, global survey of clinicians prescribing allergen immunotherapy. Expert Rev Clin Immunol. 2017;13:283-9.
4. Kutukculer N, Karaca NE, Demircioglu O, Aksu G. Increases in serum immunoglobulins to age-related normal levels in children with IgA and/or IgG subclass deficiency. Pediatr Allergy Immunol. 2007;18:167-3.
5. Ambrosino DM, Siber GR, Chilmonczyk BA, Jernberg JB, Finberg RW. An immunodeficiency characterized by impaired antibody responses to polysaccharides. N Engl J Med. 1987;316:790-3.
6. Granoff DM, Schackelford PG, Suarez BK, Nahm MH, Cates KL, Murphy TV, et al. Haemophilus influenza type B disease in children vaccinated with type B polyssacacharide vaccine. N Engl J Med. 1986;315:1584-90.
7. Perez E, Bonilla FA, Orange JS, Ballow M. Specific antibody deficiency: controversies in diagnosis and management. Front Immunol. 2017;8:586.
8. Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, et al. Primary immunodeficiencies: 2009 update. J. Allergy Clin Immunol. 2010; 125(3):771-3.
9. Stiehm ER, Casillas AM, Finkeslstein JZ. Immunodeficiency disorders: general considerations. In: Stiehm ER, Ochs HD, Winkelstein JA. Immunological disorders in infants and children. 5ª ed. Philadelphia: Elsevier Saunders; 2004. p. 289-355.
10. Boyle J. Population prevalence of diagnosed primary immunodeficiency in the US. J Clin Immunol. 2007;5(27):497-502.
11. Bousfiha AA, Jeddane L, Ailal F, Benhsaien I, Mahlaoui N, Casanova JL, Abel L. Primary immunodeficiency diseases worldwide: more common than generally thought. J Clin Immunol. 2013;33(1):1-7.
12. Mahlaoui N, Gathmann B, Kindle G, Ehl S, and the ESID Society. The European Society for Immunodeficiencies (ESID) Registry: recent advancements in the epidemiology of Primary Immunodeficiencies and how does that translate in clinical care. Int J Public Health. 2014;1(S4):25-7.
13. Leiva LE, Zelasco M, Oleastro M, Carneiro-Sampaio M, Condino- Neto A, Costa-Carvalho BT, et al. Primary immunodeficiency disease in Latin America: the second report of the LAGID directory. J Clin Immunol. 2007;27(1):101-8.
14. Roxo-Júnior P. Imunodeficiências primárias: aspectos relevantes para o pneumologista. J Bras Pneumol. 2009;35:1008-17.
15. Orange JS, Seeborg FO, Marcia B, Scalchunes C, Hernandez-Trujillo V. Family physician perspectives on primary immunodeficiency diseases. Front Med. 2016;3:12.
16. Serpa FS, Guidacci AF, Rubini NP. O atendimento médico de pacientes com doenças imunoalérgicas no Brasil: reflexões e propostas para a melhoria. Arq Asma Alerg Imunol. 2017;1(4):327-34.
17. Schatorjé EJH. The challenge of immunoglobulin-G subclass deficiency and specific polysaccharide antibody deficiency - a dutch pediatric cohort study. J Clin Immunol. 2016;36(2):141-8.
18. Errante PR, Franco JL, Espinosa-Rosales FJ, Sorensen R, Condino- Neto A. Advances in primary immunodeficiency diseases in Latin America: epidemiology, research, and perspectives. Ann N Y Acad Sci. 2012;1250:62-72.
19. LaCross NC, Marrs CF, Gilsdorf JR. Population structure in nontypeable Haemophilus influenzae. Infect Genet Evol. 2013;14:125-36.
20. Janeway CA, Travers P, Walport Mark, Shlomchik M. Imunidade adaptativa contra a infecção. In: Janeway CA, Travers P, Walport Mark, Shlomchik M. Imunobiologia - o sistema imune na saúde e na doença. 6ª ed. Porto Alegre: Artmed; 2007. p. 409-59.
21. Forte WCN. Fagócitos. In: Forte WCN. Imunologia do básico ao aplicado. 3ª ed. São Paulo: Editora Atheneu; 2015. p. 15-24.
22. Weintraub A. Immunology of bacterial polysaccharide antigens. Carbohydr Res. 2003;338(23):2539-47.
23. Obukhanych TV, Nussenzweig MC. T-independent type II immune responses generate memory B cells. J Exp Med. 2006;203(2):305-10.
24. Hyams C, Camberlein E, Cohen JM, Bax K, Brown JS. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect Immun. 2010;78:704-15.
25. Forte WCN. Sistema complemento. In: Forte WCN. Imunologia do básico ao aplicado. 3ª ed. São Paulo: Editora Atheneu; 2015. p. 25-34.
26. Ferreyra PN, Costa-Carvalho B, Orii N, Carneiro-Sampaio MMS. A síndrome de deficiência de anticorpos com imunoglobulinas normais não está relacionada ao aumento de número de células B CD19+CD5+. Rev bras alerg imunopatol. 2004;27(2):36-46.
27. Forte WCN. Imunoglobulinas. In: Forte WCN. Imunologia do básico ao aplicado. 3ª ed. São Paulo: Editora Atheneu; 2015. p. 51-64.
28. Carr TF, Koterba AP, Chandra R, Grammer LC, Conley DB, Harris KE, et al. Characterization of specific antibody deficiency in adults with medically refractory chronic rhinosinusitis. Am J Rhinol Allergy. 2011;25(4):241-4.
29. Kashani S, Carr TF, Grammer LC, Schleimer RP, Hulse KE, Kato A, et al. Clinical characteristics of adults with chronic rhinosinusitis and specific antibody deficiency. J Allergy Clin Immunol Pract. 2015;3(2):236-42.
30. Ocampo CJ, Peters AT. Antibody deficiency in chronic rhinosinusitis: epidemiology and burden of illness. Am J Rhinol Allergy. 2013;27(1):34-8.
31. Kadioglu A, Weiser JN, Paton JC, Andrew PW. The role of Streptococcus pneumoniae virulence factors in host respiratory colonization and disease. Nat Pub Group Microbiol. 2008;6:288-301.
32. Bossuyt X, Moens L, van Hoeyveld E, Jeurissen A, Bogaert G, Sauer K, et al. Coexistence of (partial) immune defects and risk of recurrent respiratory infections. Clin Chem. 2007;53:124-30.
33. Cheng YK, Decker PA, O'Byrne MM, Weiler CR. Clinical and laboratory characteristics of 75 patients with specific polysaccharide antibody deficiency syndrome. Ann Allergy Asthma Immunol. 2006;97(3):306-11.
34. Condino-Neto A, Franco JL, Trujillo-Vargas C, Espinosa-Rosales FJ, Leiva LE, Rodriguez-Quiroz F, et al. Critical issues and needs in management of Primary Immunodeficiency diseases in Latin America. Allergol Immunopathol. 2011;3(1):45-51.
35. Stiehm RS. The four most common pediatric immunodeficiencies. J Immunotoxicol. 2008;5(2):227-34.
36. Sorensen RU, Leiva LE. Measurement of pneumococcal polysaccharide antibodies. J Clin Immunol. 2014;34(2):127-8.
37. Yoshioka CRM, Martinez MB, Brandileone MCC, Ragazzi SB, Guerra MLLS, Santos SR, et al. Análise de cepas de Streptococcus pneumoniae causadores de pneumonia invasiva: sorotipos e sensibilidade aos antimicrobianos. J Ped (Rio J). 2011;87:70-5.
38. Brazilian Group for Immunodeficiency [Internet]. Sinais de alerta para Imunodeficiência primária na criança e no adulto. Disponível em: http://www. bragid. org. br. Acessado em: 27/01/2018.
39. Beppu APK, Melardi JW, Gaudino VRR, Marina Santos MC, Menezes MCS, Forte WCN. Pneumonias em imunodeficiência comum variável após mudança de ambiente físico. J Allergy Immunol. 2015;3(3):93-8.
40. Divino PHA, Basilio JHC, Fabbri RMA, Polonio IB, Forte WCN. Bronquiectasia por imunodeficiência comum variável. J Bras Pneumol. 2015;41(5):482-4. 41.
Vendrell M, de Gracia J, Rodrigo MJ, Cruz MJ, Alvarez A, Garcia M, et al. Antibody production deficiency with normal IgG levels in bronchiectasis of unknown etiology. Chest. 2005;127(1):197-204.
42. Costa-Carvalho BT, Wandalsen GF, Pulici G, Aranda CS, Solé D. Pulmonary complications in patients with antibody deficiency. Allergol Immunopathol. 2011;39(3):128-32.
43. Wall LA, Dimitriades VR, Sorensen RU. Specific antibody deficiencies. Immunol Allergy Clin North Am. 2015;35(4):659-70.
44. Boyle RJ, Le C, Balloch A, Tang ML. The clinical syndrome of specific antibody deficiency in children. Clin Exp Immunol. 2006;146(3):486-92.
45. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015;136(5):1186-205.
46. Parker AR, Allen S, Harding S. Concentration of anti-pneumococcal capsular polysaccharide IgM, IgG and IgA specific antibodies in adult blood donos. Pract Labor Med. 2016;5:1-5.
47. Borgers H, Jeruissen A, Flamaing J, Peetermans WE, Moens L, Verhaegen J, et al. Elderly subjects do not show impaired pneumococcal capsular polysaccharide serotype-specific antibody responses as assessed by a multiplexed bead assay. Clin Immunol. 2010;135:501-2.
48. Forte WCN. Imunodeficiências primárias. In: Forte WCN. Imunologia do básico ao aplicado. 3ª ed. São Paulo: Editora Atheneu; 2015. p. 221-48.
49. Paris K, Sorensen RU. Assessment and clinical interpretation of polysaccharide antibody responses. Ann Allergy Asthma Immunol. 2007;99(5):462-4.
50. Orange JS, Ballow M, Stiehm ER, Ballas ZK, Chinen J, De La Morena M, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the Basic and Clinical Immunology Interest Section of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2012 Sep;130(3 Suppl):S1-24.
51. Borgers H, Moens L, Picard C, Jeurissen A, Raes M, Sauer K, et al. Laboratory diagnosis of specific antibody deficiency to pneumococcal capsular polysaccharide antigens by multiplexed bead assay. Clin Immunol. 2010;134(2):198-205.
52. Moens L, Picard C, Shahrooei M, Wuyts G, Liston A, Fischer A, Bossuyt X. Different immunological pathways underlie the immune response to pneumococcal polysaccharides. J Clin Immunol. 2017;37(3):277-8.
53. Grumach AS. Imunodeficiências predominantemente humorais. In: Grumach AS. Alergia e imunologia na infância e na adolescência. 2ª ed. São Paulo: Editora Atheneu; 2009. p. 533-56.
54. Roux A, Schmole-Thoma B, Siber G, Hackell J, Kuhnke A, Ahlers N, et al. Comparison of pneumococcal conjugate polysaccharide and free polysaccharide vaccines in elderly adults: conjugate vaccine elicits improved antibacterial immune responses and immunological memory. Clin Infect Dis. 2008;46:1015-23.
55. Turner AEB, Gerson JE, So HY, Krasznai DJ, Hilaire AJ, Gerson DF. Novel polysaccharide-protein conjugates provide an immunogenic 13-valent pneumococcal conjugate vaccine for S. pneumoniae. Synth Syst Biotechnol. 2017;2(1):49-58.
56. Rose MA, Schubert R, Strnad N, Zielen S. Priming of immunological memory by pneumococcal conjugate vaccine in children unresponsive to 23-valent polysaccharide pneumococcal vaccine. Clin Diagn Lab Immunol. 2005;12(10):1216-22.
57. Daniels CC, Rogers PD, Shelton CM. A review of pneumococcal vaccines: current polysaccharide vaccine recommendations and future protein antigens. J Pediatr Pharmacol Ther. 2016;21(1):27-35.
58. Balloch A, Licciardi PV, Russell FM, Mulholland EK, Tang ML. Infants aged 12 months can mount adequate serotype-specific IgG responses to pneumococcal polysaccharide vaccine. J Allergy Clin Immunol. 2010;126(2):395-7.
59. Schütz K, Hughes RG, Parker A, Quinti I, Thon V, Cavaliere M, et al. Kinetics of IgM and IgA antibody response to 23-valent pneumococcal polysaccharide vaccinations in healthy subjects. J Clin Immunol. 2012;33(1):288-96.
60. Daly TM, Hill HR. Use and clinical interpretation of pneumococcal antibody measurements in the evaluation of humoral immune function. Clin Vaccine Immunol. 2015;22(2):48-152.
61. Klein DL, Martinez JE, Hickey MH, Hassouna F, Zaman K, Steinhoff M. Development and characterization of a multiplex bead-based immunoassay to quantify pneumococcal capsular polysaccharidespecific antibodies. Clin Vaccine Immunol. 2012;19(8):1276-82.
62. Elberse KE, Tcherniaeva I, Berbers GA, Schouls LM. Optimization and application of a multiplex bead-based assay to quantify serotypespecific IgG against Streptococcus pneumoniae polysaccharides: response to the booster vaccine after immunization with the pneumococcal 7-valent conjugate vaccine. Clin Vaccine Immunol. 2010;17(4):674-82.
63. Beck SC. Making sense of serotype-specific pneumococcal antibody measurements. Ann Clin Biochem. 2013;50(6):517-9.
64. Cavaliere FM, Milito C, Martini H, Schlesier M, Dräger R, Schütz K, et al. Quantification of IgM and IgA anti-pneumococcal capsular polysaccharides by a New ELISA Assay: a valuable diagnostic and prognostic tool for Common Variable Immunodeficiency. J Clin Immunol. 2013;33(4):838-46.
65. Barreto BAP, Sarinho ESC, Stefani GM, Chong-Neto HJ, Chiabai J. Alonso MLO, et al. Deficiência específica de anticorpo antipolissacarídeo de pneumococo e resposta humoral a vacinas pneumocócicas: atualização em diagnóstico. Arq Asma Alerg Imunol. 2014;1(50):253-60.
66. Goudouris, ES, Silva AMR, Ouricuri AL, Grumach AS, Condino A, Costa-Carvaho BT, e Grupo Brasileiro de IDPs da ASBAI. II Consenso Brasileiro sobre o uso de imunoglobulina humana em pacientes com imunodeficiências primárias. Einstein. 2017;15(1):1-16.
67. Fried AJ, Bonilla FA. Pathogenesis, diagnosis, and management of primary antibody deficiencies and infections. Clin Microbiol Ver. 2009;22(3):396-414.
68. Segundo GRS, Fernandes KP. Deficiência de anticorpos antipolissacarídicos: relato de casos. Rev bras alerg imunopatol. 2009;32(5):194-8.
69. García JM, Español T, Gurbindo MD, Casas C C. Update on the treatment of primary immunodeficiencies. Allergol Immunopatol. 2007;35(5):184-92.
70. Wood P. Immunotherapy for primary immunodeficiency diseases. Med Clin North Am. 2012;96(3):433-54.
71. Shehata N, Palda V, Bowen T, Haddad E, Issekutz TB, Mazer B, et al. The use of immunoglobulin therapy for patients with primary immune deficiency: an evidence-based practice guideline. Transfus Med Rev. 2010;24(1):S28-50.
72. Gardulf A, Nicolay U, Math D, Asensio O, Bernatowska E, Böck A, et al. Children and adults with primary antibody deficiencies gain quality of life by subcutaneous IgG self-infusions at home. J Allergy Clin Immunol. 2004;114(4):936-42.
73. Goudouris ES, Silva AMR, Ouricuri AL, Grumach AS, Condino A, Costa-Carvalho BT, et al. Imunoglobulina humana por via subcutânea para imunodeficiências primárias. Braz J Allergy Immunol. 2014;2(4):127-8.
74. Leiva LE, Monjure H, Sorensen RU. Modulatory role of intravenous gammaglobulin (IgIV) on the in vitro antibody response to a pneumococcal polysaccharide antigen. J Clin Immunol. 2015;35(2):206-12.
75. Sewell WA, Kerr J, Behr-Gross ME, Peter HH. Kreuth Ig Working Group. European consensus proposal for immunoglobulin therapies. Eur J Immunol. 2014;44(8):2207-14.
76. Shabaninejad H, Asgharzadeh A, Rezaei N, Rezapoor A. A comparative study of intravenous immunoglobulin and subcutaneous immunoglobulin in adult patients with Primary Immunodeficiency Diseases: a systematic review and meta-analysis. Expert Rev Clin Immunol. 2016;12(5):595-602.
77. Misbah S, Sturzenegger MH, Borte M, Shapiro RS, Wasserman RL, Berger M, Ochs HD. Subcutaneous immunoglobulin: opportunities and outlook. Clin Exp Immunol. 2009;158(1):51-9.
78. Skoda-Smith S, Torgerson TR, Ochs HD. Subcutaneous immunoglobulin replacement therapy in the treatment of patients with primary immunodeficiency disease. Ther Clin Risk Manag. 2010;6:1-10.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888