Número Atual: Outubro-Dezembro 2017 - Volume 1 - Número 4
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Síndrome de Wiskott-Aldrich com plaquetas de volume normal
Wiskott-Aldrich syndrome with normal-sized platelets
Danddara Morena Gonçalves Silveira1; Sarah Angelica Maia1; Camila Forestiero2; Gesmar Rodrigues Silva Segundo3; Débora Carla Chong-Silva4; Herberto Jose Chong Neto4; Carlos Antônio Riedi4; Troy R. Torgerson5; Nelson Augusto Rosário6
DOI: 10.5935/2526-5393.20170062
1. Residente do Serviço de Alergia e Imunologia Pediátrica do Complexo Hospital de Clínicas, Universidade Federal do Paraná (UFPR)
2. Especialista em Alergia e Imunologia pela Associaçao Brasileira de Alergia e Imunologia
3. Professor Adjunto de Pediatria, Universidade Federal de Uberlândia. Pesquisador associado do Departamento de Pediatria da University of Washington/ Seattle Children's Hospital and Research Institute
4. Professor Adjunto de Pediatria, UFPR
5. Professor do Depatamento de Pediatria, University of Washington/Seattle Children's Hospital and Research Institute. 6. Professor Titular de Pediatria, UFPR
Endereço para correspondência:
Danddara M. G. Silveira
E-mail: danddara.mgs@gmail.com
Submetido em: 12/04/2017
Aceito em: 21/06/2017
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
A Síndrome de Wiskott-Aldrich (WAS) é uma imunodeficiência congênita ligada ao cromossomo X, caracterizada por mutaçoes no gene WAS, responsável pela proteína WASP. As principais manifestaçoes clínicas sao trombocitopenia com plaquetas de volume reduzido, eczema, infecçoes recorrentes e maior incidência de doenças autoimunes e neoplasias. Relatamos o caso de um paciente do sexo masculino com sintomas clássicos desta síndrome (eczema, trombocitopenia e infecçoes recorrentes), porém com plaquetas de volume normal. Existem poucos relatos desta síndrome em pacientes com plaquetas de volume normal, o que atrasou o encaminhamento do paciente ao imunologista, o qual foi tratado como portador de Síndrome de Evans e dermatite atópica até os quatro anos de idade. A confirmaçao diagnóstica foi por teste genético. O diagnóstico precoce possibilita profilaxia com antibioticoterapia e uso de imunoglobulina endovenosa, devido ao risco de infecçoes graves, e encaminhamento para transplante de células-tronco hematopoiéticas, que até o momento é o único tratamento curativo. A suspeita clínica deve existir em pacientes com trombocitopenia inexplicável, mesmo se as plaquetas tiverem o tamanho normal, associada às outras manifestaçoes da doença.
Descritores: Síndrome de Wiskott-Aldrich, imunodeficiência primária, criança, eczema, trombocitopenia.
INTRODUÇAO
A síndrome de Wiskott-Aldrich (WAS) é uma imunodeficiência primária ligada ao X caracterizada por uma tríade clássica – imunodeficiência, eczema grave e microtrombocitopenia, além de risco aumentado para doenças autoimunes e neoplasias associadas1. Em 1937, Alfred Wiskott, pediatra alemao, relatou o caso de três irmaos com trombocitopenia, disenteria, eczema e otites de repetiçao que faleceram antes dos dois anos de idade por sangramento intestinal e sepse. Em 1954, Robert Aldrich e seus colaboradores relataram o caso de uma família com 16 homens de 6 geraçoes que faleceram por doença que apresentava as mesmas características descritas por Wiskott, identificando um padrao de herança ligada ao X. Após 40 anos, em 1994, o gene responsável pela doença foi identificado no braço curto do cromossoma X, na posiçao Xp11.22-p11.231-3.
Estima-se que a incidência da doença nos Estados Unidos da América seja de aproximadamente 1 a 4 casos por 1.000.000 meninos nascidos vivos, e represente 1,2% das imunodeficiências primárias relatadas no país. A média de idade para o diagnóstico é de 24 meses em famílias sem casos prévios. Casos raros foram descritos em meninas, envolvendo uma mutaçao deletéria do cromossomo X derivado do pai, e inativaçao nao randômica do cromossomo X, derivado da mae4.
O gene responsável pela WAS codifica a proteína WASp, que contém 502 aminoácidos, e é expressa no citoplasma de células hematopoiéticas nao eritroides, incluindo as células tronco (CD34+), plaquetas, macrófagos, células dendríticas, linfócitos e neutrófilos. A proteína WASp regula a sinapse imune, a locomoçao e sinalizaçao celular, e participa do processo de fagocitose e de apoptose. O gene WAS pode apresentar mutaçoes heterogêneas, que vao estar relacionadas com o espectro da doença. Dependendo do tipo da mutaçao, a funçao e/ou expressao intracelular de WASp é afetada de forma diferente. A mutaçao genética de WAS pode se apresentar como a clássica Síndrome de Wiskott-Aldrich e suas variaçoes fenotípicas, Trombocitopenia ligada ao X, ou Neutropenia ligada ao X4-6.
Em geral, mutaçoes que levam a níveis reduzidos de WASp resultam em trombocitopenia ligada ao X, enquanto que as mutaçoes que abolem a expressao da WASp ou resultam na expressao de uma proteína truncada estao associadas com a síndrome7. Utilizase ainda um sistema de pontuaçao para classificar a gravidade da doença. Pontuaçao de 1 indica que o paciente tem apenas trombocitopenia; pontuaçao de 2 quando o paciente apresenta trombocitopenia com achados adicionais, como eczema leve ou transitório ou infecçoes brandas. Pacientes com pontuaçao de 1 e 2 sao designados como tendo Trombocitopenia ligada ao X. Aqueles com eczema resistente ao tratamento e infecçoes recorrentes, recebem pontuaçao de 3 (WAS leve) ou 4 (WAS grave). A pontuaçao de 5 é dada a pacientes com doença autoimune ou malignidade, independentemente do escore original8,9.
Na WAS há comprometimento da imunidade humoral, celular e inata. Há níveis séricos reduzidos de IgG, IgM e IgA e níveis aumentados de IgE, além de respostas anormais de anticorpos funcionais, como isohemaglutininas e de anticorpos vacinais, como tétano, pneumococco e vacinas conjugadas. Os pacientes apresentam, ainda, defeitos quantitativos e qualitativos nas células Th2, com linfopenia, porém com contagem de linfócitos > 1,000/μL na maioria dos pacientes. Células NK, monócitos e neutrófilos estao em número normal, porém com deficiência funcional. Há susceptibilidade aumentada para infecçoes sinusopulmonares, principalmente pneumonias e otites, sepse e meningite em menor número, além de infecçoes de pele como impetigo e celulite. Infecçoes atípicas, como varicela grave, Herpes simplex, citomegalovírus, vírus Epstein-barr induzindo distúrbios mieloproliferativos e pneumonia por Pneumocystis jiroveci, também acometem os pacientes1,3,4.
A trombocitopenia é quase universal nos pacientes com WAS. As plaquetas sao destruídas no baço por apresentarem alteraçoes morfológicas e funcionais, incluindo a falta de grânulos específicos, número reduzido de organelas no citoplasma e defeitos de agregaçao. Metade dos pacientes tem plaquetopenia < 20.000/μL. Complicaçoes hemorrágicas ocorrem em 80% dos pacientes, sendo as mais frequentes: epistaxe, equimoses, petéquias, hemorragia gastrointestinal e hemorragia intracraniana1,4,6.
Autoimunidade e neoplasias podem estar associadas a WAS. Doenças autoimunes sao comuns na WAS e podem acometer até 40% dos pacientes, além de serem preditoras do desenvolvimento de malignidades posteriormente4. A autoimunidade no cenário de WAS pode ser devida à formaçao de autoanticorpos ou à presença de clones de células T autorreativos. As manifestaçoes mais comuns sao anemia hemolítica autoimune, neutropenia autoimune, vasculite, nefropatia por IgA, artrites e doença inflamatória intestinal3,4,7.
O desenvolvimento de malignidade é frequente, com prevalência estimada de 13-20% dos pacientes, com uma idade mediana de início de 9,5 anos. Linfoma é a neoplasia mais comum, com predomínio do Linfoma nao Hodgkin, seguido por Mielodisplasia, Leucemia e Distúrbios mieloproliferativos3,4,10.
DIAGNOSTICO
A seguir, sao descritos os critérios diagnósticos, de acordo com a Sociedade Europeia de Imunodeficiência11.
Definitivo
Paciente do sexo masculino com trombocitopenia congênita (menos de 70.000 plaquetas/mm3), plaquetas pequenas e pelo menos uma das seguintes alteraçoes:
– Mutaçao na proteína WAS (WASP);
– Ausência de mRNA da WASP em análise "Northern blot" de linfócitos;
– Ausência de proteína WASP em linfócitos;
– Primos maternos, tios ou sobrinhos com plaquetas pequenas e trombocitopenia.
Provável
Paciente do sexo masculino com trombocitopenia congênita (menos de 70.000 plaquetas/mm3), plaquetas pequenas e pelo menos uma das seguintes alteraçoes:
– Eczema;
– Resposta anormal de anticorpos a antígenos polissacarídicos;
– Infecçoes bacterianas ou virais;
– Doenças autoimunes;
– Linfoma, leucemia ou tumor cerebral.
Possível
Paciente do sexo masculino com trombocitopenia (menos de 70.000 plaquetas/mm3) e plaquetas pequenas; ou paciente do sexo masculino esplenectomizado por trombocitopenia, que tem pelo menos uma das seguintes alteraçoes:
– Eczema;
– Resposta anormal de anticorpos a antígenos polissacarídicos;
– Infecçoes bacterianas ou virais recorrentes;
– Doenças autoimunes;
– Linfoma, leucemia ou tumor no cérebro.
TRATAMENTO
O tratamento de suporte consiste na reposiçao de imunoglobulina endovenosa, uso de sulfametoxazol + trimetoprim para prevençao de pneumonia por Pneumocystis jiroveci, antibióticos profiláticos para infecçoes sinusopulmonares, uso de aciclovir para casos de infecçoes herpéticas, transfusoes de plaquetas para casos de sangramentos e medicaçoes tópicas para controle do eczema. A esplenectomia nao é recomendada pelo risco de infecçoes. Até o momento, o único tratamento curativo é o transplante de células tronco hematopoiéticas1,4,6,12.
RELATO DE CASO
L.B.C., sexo masculino, 4 anos. Aos 2 meses de idade apresentou anemia importante associada a trombocitopenia, verificado em exame de puericultura. Nessa ocasiao, necessitou internaçao hospitalar e múltiplas transfusoes de sangue, sendo entao encaminhado para avaliaçao no ambulatório de hematologia pediátrica. Devido à suspeita de síndrome de Evans (anemia hemolítica e trombocitopenia), recebeu tratamento com corticoterapia sistêmica, com pouca melhora do quadro.
Aos 4 meses, paciente iniciou com lesoes eczematosas em face, com posterior progressao para todo o corpo, muito pruriginosas. Encaminhado para avaliaçao com dermatologista, recebeu diagnóstico de dermatite atópica, mas as lesoes nao responderam ao tratamento clínico (Figura 1).
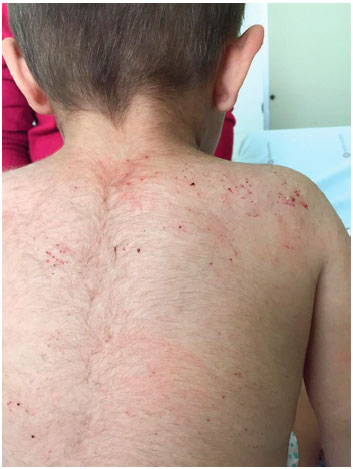
Figura 1 Lesoes eczematosas em regiao dorsal
Aos 6 meses, apresentou edema em joelho direito, sinais flogísticos, além de petéquias em membros inferiores. Na ocasiao, apresentava trombocitopenia (33.000 plaquetas), sendo optado por internaçao hospitalar para a realizaçao de pulsoterapia com corticoide. Aos 7 meses e aos 8 meses apresentou novos episódios de edema em joelhos, sem sinais flogísticos, com melhora espontânea. Aos 10 meses, apresentou dois episódios de edema em pé esquerdo, associados a dor e calor local, com melhora após uso de paracetamol.
Com 1 ano e 10 meses, paciente necessitou de internaçao hospitalar por sepse de foco pulmonar. Durante o internamento, desenvolveu quadro de lesoes cutâneas, cuja biópsia foi compatível com diagnóstico de vasculite (variante de Púrpura de Henoch-Schönlein). Com 2 anos e 5 meses, foi realizada uma nova biópsia cutânea em lesao bolhosa em pé esquerdo, com resultado também compatível com vasculite.
Com 2 anos e 7 meses, o paciente apresentou hemorragia digestiva alta, com necessidade de internaçao hospitalar e transfusao de plaquetas. Até os 3 anos de idade, paciente apresentou outros quatro internamentos por infecçoes, sendo um devido a infecçao de cateter venoso central com hemocultura positiva para leveduras e boa resposta a tratamento utilizando micafungina, e três internamentos devido a pneumonias com derrame pleural. Todos os internamentos foram por intercorrências graves, com necessidade de internaçao em UTI pediátrica e tratamento agressivo com antibioticoterapia. Após apresentar quadro de edema em ombros e joelhos durante uma das internaçoes por pneumonia, foi avaliado por reumatologista, com posterior diagnóstico de lupus eritematoso sistêmico.
Devido ao quadro frequente de infecçoes graves, paciente foi referenciado para avaliaçao no serviço de imunologia pediátrica. Foram revisados os hemogramas anteriores e em todos o volume plaquetário médio (VPM) encontrava-se na faixa da normalidade ou próximo a ela. Solicitados novos exames demonstrando VPM de 9,8 fl (referência 9,4-12,6 fl), ausência de resposta após vacinaçao com Pneumo 23®, isohemaglutininas baixas (tipo sanguíneo O+, com anti-A reagente 1:4 e anti-B nao reagente), ausência de soroconversao após vacinaçao para Hepatite B, IgE e IgG elevadas (> 1000 Ku/L e 578,9 Ku/L, respectivamente) e linfopenia acentuada de células T. A pesquisa de mutaçao do gene WAS foi realizada pelo Immunology Diagnostic Laboratory do Seattle Children's Hospital and Research Institute que verificou a presença da mutaçao c.354delT no exon 3 causando uma alteraçao do tipo frameshift e uma parada prematura de codificaçao (stop-codon) 7 codons adiante (p.G119Efs*), que prediz a ausência da proteína WASp e um grave fenótipo clínico.
Após diagnóstico de síndrome de Wiskott-Aldrich, o paciente iniciou tratamento com imunoglobulina humana endovenosa mensal, além de profilaxia com sulfametoxazol-trimetoprim e foi encaminhado para o serviço de Transplante de Medula Ossea. Recebeu transplante haploidêntico de origem paterna, e no momento encontra-se em recuperaçao.
DISCUSSAO
Classicamente, a síndrome de Wiskott-Aldrich caracteriza-se por ser uma imunodeficiência com trombocitopenia e com microplaquetas. Entretanto, relatamos um paciente do sexo masculino com sintomas clássicos desta síndrome (eczema, trombocitopenia e tendência a infecçoes recorrentes), porém com plaquetas de volume normal.
Existem poucos relatos desta síndrome em pacientes com plaquetas de volume normal. Em 2014, Mantakadis et al. relataram 3 casos de pacientes portadores dessa síndrome que nao apresentavam alteraçoes no volume das plaquetas.Todos os pacientes apresentavam apenas trombocitopenia, sem eczema ou tendência aumentada a infecçoes ou doenças autoimunes. Destes, dois pacientes apresentaram a mutaçao c.854_855 no exon 9, enquanto o terceiro paciente apresentou a mutaçao c.743_743+1delAG no exon 713. Baharin et al. apresentaram um caso de um bebê apresentando todas as características clássicas da síndrome, exceto por plaquetas cujos volumes eram normais, e cuja mutaçao era c.1264G>T, no exon 108. Patel et al. relataram um caso similar de paciente expressando a forma clássica da síndrome e sem alteraçoes no volume das plaquetas, demonstrando a mutaçao c.862A>T no exon 914. O paciente deste relato apresentou a mutaçao c.354delT no exon 3, diferente de todas as demais relatadas.
Atualmente, existem mais de 150 mutaçoes diferentes do gene WAS relatadas, com diferentes expressoes sobre a proteína da WAS e, consequentemente, diferentes fenótipos clínicos9. Uma possibilidade é que, embora rara, a manifestaçao de WAS com plaquetas normais seja um novo fenótipo da doença14.
Pela associaçao da síndrome de Wiskott-Aldrich com quadro de lupus eritematoso sistêmico, nosso paciente enquadra-se na classe 5 de gravidade da doença (SWA associada com doença autoimune ou doença neoplásica). O paciente relatado teve seu diagnóstico firmado apenas aos quatro anos, inicialmente tratado como Síndrome de Evans e como dermatite atópica. A síndrome de Evans foi suspeitada por o paciente apresentar anemia hemolítica associada à plaquetopenia. Frequentemente, casos mais leves de SWA apresentam apenas trombocitopenia congênita, e sao tratados como portadores de trombocitopenia imune primária, o que atrasa o tratamento adequado.
A síndrome de Wiskott-Aldrich é uma imunodeficiência com múltiplos fenótipos e associada a risco de desenvolvimento posterior de neoplasias e doenças autoimunes. O diagnóstico precoce é importante para adequado tratamento da doença, com possibilidade de cura com transplante de medula óssea. Apesar da tríade clássica envolver pacientes do sexo masculino com microtrombocitopenia, eczema e propensao a infecçoes de repetiçao, a suspeita clínica deve existir caso o paciente apresente trombocitopenia inexplicável, mesmo quando as plaquetas tiverem o tamanho normal, principalmente se o paciente apresentar outras manifestaçoes clássicas da doença8,14.
REFERENCIAS
1. Ariga T. Wiskott-Aldrich syndrome; an X-linked primary immunodeficiency disease with unique and characteristic features. Allergol Int. 2012;61:183-9.
2. Notarangelo LD, Miao CH, Ochs HD. Wiskott-Aldrich Syndrome. Curr Opin Hematol. 2008;15:30-6.
3. Worth AJ, Thrasher AJ. Current and emerging treatment options for Wiskott-Aldrich syndrome. Expert Rev Clin Immunol. 2015;11:1015-32.
4. Buchbinder D, Nugent DJ, Fillipovich AH. Wiskott-Aldrich syndrome: diagnosis, current management, and emerging treatments. Appl Clin Genet. 2014;7:55-66.
5. Sasahara Y. WASP-WIP complex in the molecular pathogenesis of Wiskott-Aldrich syndrome. Pediatr Int. 2016;58:4-7.
6. Gonzalez IG, Carvalho BTC. Síndrome de Wiskott-Aldrich. Rev bras alerg imunopatol. 2011;34:59-64.
7. Massaad MJ, Ramesh N, Geha RS. Wiskott-Aldrich syndrome: a comprehensive review. Ann N Y Acad Sci. 2013;1285:26-43.
8. Baharin MF, Dhaliwal JS, Sarachandran SV V., Idris SZ, Yeoh SL. A rare case of Wiskott-Aldrich Syndrome with normal platelet size: a case report. J Med Case Rep. 2016;10:188.
9. Albert MH, Notarangelo LD, Ochs HD. Clinical spectrum, pathophysiology and treatment of the Wiskott-Aldrich syndrome. Curr Opin Hematol. 2011;18:42-8.
10. Mortaz E, Tabarsi P, Mansouri D, Khosravi A, Garssen J, Velayati A, et al. Cancers related to immunodeficiencies: Update and perspectives. Front Immunol. 2016;7:1-13.
11. Guzman D, Veit D, Knerr V, Kindle G, Gathmann B, Eades-Perner AM, et al. The ESID Online Database network. Bioinformatics. 2007;23:654-5.
12. Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott- Aldrich Syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1 Suppl.):84-90.
13. Mantadakis E, Sawalle-Belohradsky J, Tzanoudaki M, Kanariou M, Chatzimichael A, Albert MH. X-linked thrombocytopenia in three males with normal sized platelets due to novel WAS gene mutations. Pediatr Blood Cancer. 2014;61:2305-6.
14. Patel PD, Samanich JM, Mitchell WB, Manwani D. A unique presentation of Wiskott-Aldrich syndrome in relation to platelet size. Pediatr Blood Cancer. 2011;56:1127-29.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888