Número Atual: Julho-Setembro 2017 - Volume 1 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
Uma nova classe de doenças: doenças autoinflamatórias
A new class of diseases: autoinflammatory diseases
Leonardo Oliveira Mendonça1; Ricardo Krieger Azzolini2; Joao Paulo de Assis3; André Franco4; Jorge Kalil5; Fabio Morato Castro6; Alessandra Pontillo7; Myrthes Toledo de Barros8
DOI: 10.5935/2526-5393.20170037
1. Médico Imunologista e Alergista do Serviço de Imunologia Clínica e Alergia, Hospital das Clínicas, Faculdade de Medicina da Universidade de Sao Paulo (HC-FMUSP)
2. Médico Reumatologista do Serviço de Reumatologia, HC-FMUSP
3. Médico Residente de Clínica Médica, Hospital Albert Einstein
4. Acadêmico - Aluno de graduaçao da Faculdade de Medicina da USP (FM-USP)
5. Professor Titular da Disciplina de Imunologia Clínica e Alergia, FM-USP
6. Professor Associado da Disciplina de Imunologia Clínica e Alergia, HC-FMUSP
7. Professora Titular Faculdade de Biomedicina da USP
8. Médica Assistente do Serviço de Imunologia Clínica e Alergia, HC-FMUSP
Endereço para correspondência:
Leonardo Oliveira Mendonça
E-mail: leonardo.oliveira.mendonca@gmail.com
Submetido em: 09/06/2017
Aceito em: 03/07/2017.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
As doenças autoinflamatórias sistêmicas sao um grupo de doenças raras recentemente descritas, mas que vêm ganhando espaço no cenário clínico. Caracterizam-se por alteraçoes da imunidade inata, portanto sem a presença de linfócito T autorreator ou autoanticorpo, e que respondem ao bloqueio de uma única citocina. Esta revisao tem como objetivo analisar a base imunofisiológica destas doenças e descrever brevemente cada uma delas com suas características clínicas mais importantes.
Descritores: Inflamaçao, doenças hereditárias autoinflamatórias, imunidade inata.
INTRODUÇAO
As doenças inflamatórias sistêmicas, inicialmente classificadas como autoimunes, foram redefinidas nas últimas décadas com o aprofundamento do conhecimento do sistema imunológico1. Baseando-se no compartimento imunológico envolvido, essas doenças atualmente sao classificadas em dois grandes grupos: autoinflamatórias e autoimunes. Apesar de parecer apenas definiçoes didáticas, esta diferenciaçao é imprescindível para o diagnóstico, tratamento imunológico alvo e prognóstico clínico corretos2.
Por definiçao as doenças autoimunes sao desordens sistêmicas originadas no sistema imune adaptativo. A perda da tolerância imunológica central ou periférica de linfócitos T ou B e a consequente formaçao de autoanticorpos com destruiçao tecidual anticorpo específico sao características dessas doenças3.
Por outro lado, as doenças autoinflamatórias sao doenças geradas a partir de desregulaçao na imunidade inata. Deste modo, a inexistência de linfócitos T autorreatores e de anticorpos tecido-específicos definem determinadas entidades clínicas como autoinflamatórias4,5.
Este novo grupo de doenças vem ganhando grande espaço no cenário clínico pediátrico desde a descoberta do gene responsável pela Febre Familiar do Mediterrâneo (FFM), por Daniel Kastner, em 19976. Sua classificaçao e caracterizaçao clínica sao atualmente bem estabelecidas somente na faixa etária pediátrica7. Em adultos, pouco se sabe sobre esse conjunto de doenças, seja por fenótipos diferentes, por atraso no diagnóstico, ou por manifestaçoes incompletas do esperado8,9.
As doenças autoimunes e autoinflamatórias podem ser monogênicas ou poligênicas, de herança e penetrância variáveis e podem ter origem em células germinativas ou somáticas10,11. Este estudo propoese a rever a imunologia, a fisiopatologia e as características clínicas das principais doenças monogênicas autoinflamatórias.
IMUNOLOGIA E FISIOPATOLOGIA
A imunidade inata é conhecida por ser a primeira barreira que diferencia o que é próprio do nao próprio do organismo humano12,13. A distinçao é feita através do reconhecimento de padroes pelos pattern-recognition receptors (PRRs)14. Os PRRs sao expressos por células da primeira linha de defesa, como os macrófagos, monócitos, células dendríticas, neutrófilos e células epiteliais. Entretanto, células da imunidade adaptativa também podem expressar estes receptores15,16.
Os PRRs extracelulares sao os Toll-like receptors (TLRs) e os C-typelectins (CTLs) que reconhecem os PAMPs (pathogens-associated molecular patterns) e sinalizam à célula qual tipo de organismo a está atacando17,18. Após o reconhecimento externo, outros PRRs intracelulares sao responsáveis pela diferenciaçao da resposta, principalmente os RNA-sensing, RIG like helicase (RLHs), principalmente RIG 1 e MDA5, e os DNA sensors, DAI e AIM219,20. A ativaçao de qualquer um destes receptores ativa uma via comum inflamatória que depende da degradaçao do sistema NFκβ e a produçao de citocinas pró-inflamatórias derivadas da IFN tipo 121,22.
Outra via de reconhecimento intracelular sao os NOD likere ceptors (NLRs) que reconhecem nao só os PAMPs, mas também os sinais inflamatórios nao infecciosos conhecidos como DAMPs (dangerassociated molecular patterns)23. A consequência da ativaçao desta via é a formaçao de uma plataforma dependente da caspase 1 (os "inflamassomas") que sao responsáveis pela maturaçao e liberaçao de IL 1β e IL 18, importantes citocinas inflamatórias24.
A plataforma mais famosa e bem estuda responsável pela inflamaçao nas doenças autoinflamatórias é o inflamossoma, que é constituído tipicamente por: (1) proteína temporária típica; (2) molécula adaptadora pequena ASC (apoptosis associated speck-like protein contendo um domíneo CARD); e (3) a forma progenitora da enzima caspase-1 (pró-caspase1), que participa da síntese das interleucinas inflamatórias e que consequentemente ativam as células T e B25.
Pelo menos quatro proteínas temporárias foram descritas: NLRP1 (família do NOD like receptor, com um domínio contendo a pirina) ativado pelo Bacillus anthracis; AIM2 (ausente no melanoma tipo 2) onde o inflamassoma pode ser ativado por dsDNA; NLRC4, que pertence à família do NLR CARD (caspase activation and recruitment domain), contém a proteína 4 como domínio e é ativado pela flagelina; e NLRP3 (família do NOD like receptor, com um domínio contendo a proteína 3), o inflamassoma responsável pela IL-1β26,27.
A montagem do inflamassoma é feita através da ligaçao do domínio pirina do ASC com o domínio pirina da proteína essencial. Automaticamente é recrutado um domínio de pró-caspase 1 (domínio CARD) que forma entao o complexo multiproteico. A única forma de ativar diretamente a pró-caspase 1 é através do NLRC4, que nao requer o domínio ASC28-30.
Contudo, nem tudo é mediado pela interleucina 1. A descriçao de um grupo de doenças mediadas por interferon tipo-1, as síndromes autoinflamatórias associadas ao proteossoma (PRAAS - proteasome-associated autoinflammatory syndromes) desmistificou este conceito. O proteossoma é um sistema complexo que degrada proteínas intracelulares ubiquinizadas (marcadas com uma guanina para serem eliminadas). Mutaçoes de perda de funçao no gene PSMB8, responsável pela transcriçao da subunidade β5i do proteossoma, leva ao acúmulo de proteínas ubiquinizadas no citoplasma e, portanto, desencadeia inflamaçao dependente desta via21.
Os interferons também sao moléculas pertencentes à primeira linha de defesa do corpo, a imunidade inata. A produçao de interferon é feita através de reconhecimento de padroes intracelulares no citoplasma ou endossomas, e sao reconhecidos por inúmeros sensores, tais como: TLRs, RIG-1-like, NOD-like e um grande número de sensores de DNA: AIM2, GMP-AMP sintetase (cGAS) e proteína 16 induzida por IFN-γ 22. Neste caso em particular, a ativaçao do sistema inicia-se através da interaçao do DNA dupla hélice com a enzima cGAS, que é responsável por catalisar de forma nao canônica o dinucleotídeo cíclico cGAMP. Este dinucleotídeo encontra e ativa a proteína STING, que, após ativaçao, migra do retículo endotelial para o compartimento intermediário de Golgi no citoplasma, aonde o sinal é propagado através da fosforilaçao da cinase TANK-ligadora tipo 1 (TBK1), com formaçao de uma família de citocinas conhecidas como fatores reguladores de interferon. Estes fatores migram até o núcleo e de forma autócrina induzem a síntese de interferon tipo α (IFN-α) (Figura 1). O interferon tipo 1 liga-se ao mesmo heterodímero expresso por todas as células nucleadas. Estes receptores quando ativados induzem fosforilaçao do sistema Janus Kinase (JAK), TYK2 e ativaçao de diferentes famílias de STAT22.
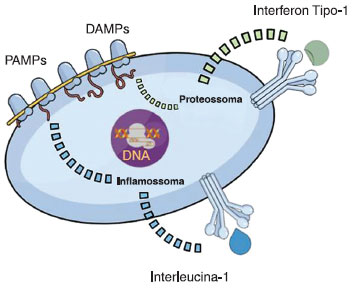
Figura 1 Vias de ativaçao da imunidade inata através dos DAMPs (danger-associated molecular patterns) e PAMPs (pattogen-associated molecular patterns) e ativaçao das vias inflamatórias na imunidade inata (inflamossoma e proteossoma)
Duas outras vias inflamatórias responsáveis por doenças autoinflamatórias foram descobertas recentemente. Uma delas baseia-se no sistema complemento e doenças mediadas por ele, sem ativaçao da imunidade adaptativa, como ocorre na síndrome hemolítica urêmica. A outra, baseia-se na degradaçao direta do sistema NF-κβ via núcleo da célula, com produçao de IL6, TNF, IL1 e IL18, a depender do estímulo21.
Em todos os casos, a liberaçao de citocinas inflamatórias recruta para o local inflamado células que causam destruiçao tecidual local, perpetuando a inflamaçao sistêmica ou local. O tipo celular, o tecido e o nível de inflamaçao dependem do agente causador e do padrao de reconhecimento, bem como da citocina liberada pelas células infiltrando o tecido alvo31.
SINDROMES CLINICAS
Febres recorrentes
Síndromes Periódicas Associadas à Criopirina - CAPS (Cryopyrin Associated Periodic Syndromes - OMIM #120100):
Este grupo de doenças está associado a mutaçoes no mesmo gene, NLRP3. A prevalência estimada desta doença é desconhecida no Brasil e no mundo, mas estima-se que na França seja de 1/360.000 mil indivíduos. Clinicamente, é uma doença espectral por gravidade inflamatória, sendo possível classificá-las em: FCAS (familial cold autoinflammatory syndrome); Muckle-Wells e NOMID/ CINCA (neonatal onset multisystemic inflamatory disease)32. Os achados comuns a este grupo de doenças sao: febre recorrente, manifestaçoes articulares (artralgia a artrite deformante) e rash cutâneo, em sua maioria semelhante à urticária. O rash urticariforme, que é o achado mais consistente entre as síndromes é migratório, maculopapular, e, geralmente, nao ou pouco pruriginoso33. Na FCAS, o quadro é precipitado por exposiçao ao frio e tende a ter menor dimensao sistêmica. A Muckle-Wells nao é precipitada pelo frio e o quadro ganha gravidade pelo fato de surdez neurosensorial ser encontrada em até 20% dos pacientes e a artrite ser mais importante, mas o acometimento do sistema nervoso central é pouco frequente34,35. Já a NOMID é grave e de início precoce na infância, que, além do quadro acima descrito, apresenta atraso no crescimento, papiledema, hidrocefalia e artrite importante e deformante. Em qualquer um dos três expectros pode também ser observado acometimento ocular, que se manifesta através de conjuntivite nao infecciosa, uveíte e iridociclite. Nao raramente, sao encontradas vasculites, osteopenia, hepatoesplenomegalia e linfonodomegalia. A amiloidose secundária é uma complicaçao grave da síndrome e este risco aumenta com a maior gravidade das doenças, sendo o risco de aproximadamente 20 a 30% na Muckle-Wells, e de 40 a 60% na NOMID36-38.
Síndrome de Schnitzler (sem registro OMIM)
A síndrome de Schnitzler é uma doença autoinflamatória complexa, geneticamente indefinida e também pode estar relacionada a mutaçoes no gene NLRP3 (aproximadamente 30 a 40% têm mutaçao neste gene)39,40. Geralmente inicia-se por erupçao cutânea do tipo macular róseo-avermelhada que pode ser acompanhada de prurido. As lesoes podem ser disseminadas, embora o envolvimento da face e das extremidades seja menos comum. A frequência e a duraçao das crises sao variáveis. Urticária induzida pelo frio pode fazer parte das manifestaçoes cutâneas41. A febre intermitente associada ao rash é comum e geralmente acima de 40 °C. Lesoes ósseas dolorosas e dor articular sao prevalentes. Os níveis de IgM podem permanecer estáveis ou aumentar progressivamente durante as crises, mas geralmente os indivíduos apresentam picos monoclonais constantes de IgM ou IgG42,43. Estao presentes: anemia secundária às crises inflamatórias, trombocitose, linfonodomegalia com características inflamatórias e hepatoesplenomegalia. A amiloidose secundária pode estar presente mas em menor gravidade, e existe risco de evoluçao para mieloma múltiplo44,45.
Febre Familiar do Mediterrâneo (OMIM #249100)
A Febre Familiar do Mediterrâneo (FFM) é uma doença induzida por defeito na proteína conhecida como pirina, e está relacionado a mutaçoes no gene MEFV (Mediterranean fever)46. A FFM é a doença autoinflamatória monogênica mais comum com prevalência global estimada de 1:50.000 indivíduos na regiao mediterrânea47,48. No Brasil ainda nao temos estimativas desta prevalência49. Clinicamente, a doença cursa com febre recorrente que dura em média entre um e três dias (38 °C - 40 °C), dor abdominal difusa ou localizada, alteraçoes do hábito intestinal, artralgia (em grandes articulaçoes), artrite (em joelhos e membros superiores/inferiores), dor torácica (pleurite) e, mais raramente, pericardite. Há envolvimento cutâneo através de lesoes eritematosas, erisipela-like ou púrpura de Henoch-Schönlein, principalmente em membros inferiores50. Estas crises podem ser precedidas por mialgia, cefaleias, náuseas e dispneia. Meningite asséptica, esplenomegalia, linfonodomegalia e poliarterite nodosa podem ocorrer. A amiloidose secundária pode ser uma complicaçao grave em longo prazo, e diversas correlaçoes fenotípicas e genotípicas já foram feitas51.
Defeito da Mevalonato Quinase (OMIM: HiperIgD #260920 / Acidúria Mevalônica #610377)
Também é um grupo de doenças espectrais relacionadas às mutaçoes do gene MVK (mevalonato kinase). Defeitos neste gene estao relacionados à Síndrome Hiper-IgD, que varia de doença mais leve e branda até doença grave com alteraçoes do SNC, surdez e até amiloidose sistêmica52,53. Elas se manifestam por episódios de febre que se intensificam com o passar do tempo, e duram em média de 3 a 7 dias54. Além de elevaçao nos marcadores de fase aguda, outras alteraçoes laboratoriais frequentemente encontradas é o aumento de níveis séricos de IgA e IgD e pode haver aumento da excreçao de ácido mevalônico. A linfadenopatia dolorosa é uma manifestaçao muito comum, afetando principalmente a cadeia cervical. Dor abdominal também é um sintoma frequente associado a vômitos e/ou diarreia. Comumente ocorre cefaleia e esplenomegalia e, menos frequentemente, hepatomegalia. Poliartralgia e artrite simétrica nao erosiva de grandes articulaçoes, especialmente joelhos e tornozelos estao presentes em uma grande parcela dos pacientes. Lesoes cutâneas difusas, maculopapulares eritematosas e urticariformes, nódulos eritematosos, petéquias ou lesoes purpúricas sao frequentes. Manifestaçoes neurológicas podem estar presentes como: microcefalia, dolicocefalia, retardo mental, ataxia e atrofia cerebelar e epilepsia, assim como manifestaçoes oculares: uveíte, catarata e esclera azulada55-58.
Síndrome Periódica Associada ao Receptor do Fator de Necrose Tumoral (TRAPS - TNF Receptor-Associated Periodic Syndrome - OMIM #142680)
Trata-se de doença relacionada a mutaçoes no gene TNFRSF1A59. Mutaçoes neste receptor, que é responsável pela sinalizaçao intracelular, causam excesso de ativaçao de vias inflamatórias cursando em aumento de secreçao de IL-1β. Os pacientes apresentam-se com febre, geralmente mais duradoura (mais de 7 dias e até 21 dias), dor abdominal súbita e de forte intensidade, que sao os principais sintomas60. Mialgia migratória também é bastante comum e pode vir acompanhada de máculas eritematosas ou, mais raramente, placas edematosas e urticariformes que migram juntamente com a dor. Podem ocorrer manifestaçoes oculares como: conjuntivite e uveíte, com dor, e hiperemia associada a edema periorbitário. Manifestaçoes no SNC incluem meningite, neurite óptica e alteraçoes comportamentais. Serosites, principalmente derrame pericárdico e pleural, sao comuns61-63.
Doenças piogênicas inflamatórias
Doenças Osseas Inflamatórias (OMIM: DIRA - Deficiência do Antagonista do Receptor de Interleucina 1 #612852 / Síndrome de Majeed #609628 /OMRC - Osteomielite Multifocal Recorrente Crônica #259680/ PAPA - Artrite Estéril Piogênica, Pioderma Gangrenoso e Acne #604416)
Este grupo de doenças caracterizam-se por osteomielite estéril associadas ou nao a manifestaçoes cutâneas, hematológicas e evoluçao clínica aguda agressiva ou crônica e lenta. Como síndrome, todas devem ser enfocadas como osteomielite multifocal. Quando de início precoce, nos primeiros meses de vida, e associada ao acometimento cutâneo com rash tipo pustuloso, predominante no dorso e na fronte, com ausência de febre e com mutaçao patogênica no gene ILR1N chama-se de DIRA (Deficiency of Interleukin 1 Receptor Antagonist). Já foi descrito diagnóstico tardio em crianças de 12 anos, mas nunca em adultos e, se nao tratada, pode levar à insuficiência respiratória e morte por tempestade de citocinas64. Já a Síndrome de Majeed, caracteriza-se por mutaçoes no gene LPIN2, localizado no cromossomo 18p11. Clinicamente manifesta-se como osteomielite crônica recorrente, associada à anemia desiridropoiética congênica (anemia caracterizada por icterícia leve ou moderada, haptoglobina baixa ou ausente, com reticulócitos elevados, nao correspondente ao nível da anemia) e dermatoses neutrofílicas que lembram síndrome de Sweet. A síndrome PAPA (Pyogenic Sterile Arthritis, Pyoderma Gangrenosum and Acne) está relacionada à mutaçao do gene PSTPIP1, localizado no cromossomo 15q24. Quase 95% dos pacientes apresentam-se com artrite piogênica estéril, sendo os achados cutâneos variáveis em gravidade e localizaçao. Versoes espectrais do pioderma podem ser encontradas como Síndromes de Sweet e Hidradenite supurativa. A acne pode se apresentar em forma de abscessos faciais ou em qualquer outro local do corpo, geralmente grave. Variantes como PASH (pioderma artrite e hidradenite supurativa), PAPASH (pioderma, acne, artrite piogênica e hidradenite) e PASS (pioderma, artrite piogênica e sacroileíte) já foram descritas e atribuídas à mesma classe de doenças65,66. Quando nao há acometimento de qualquer outro órgao e evoluçao aguda recorrente (crônica) chamamos o quadro de osteomielite multifocal recorrente crônica.
Doenças piogênicas cutâneas (OMIM: DITRA - Deficiência do Receptor de Interleucina 36 #614204 /Psoríase tipo 2 #602723)
Este grupo de doenças caracteriza-se por distúrbios cutâneos do tipo psoríase com mutaçao nos genes CARD14 e IL36RN67. Tipicamente a DITRA apresenta-se em surtos recorrentes. O quadro inicia-se de forma abrupta com: febre baixa, artrite e pústulas cutâneas generalizadas, com elevaçao de provas inflamatórias que podem evoluir para infecçao cutânea e morte por sepse. Já a psoríase tipo 2 tem o comportamento de lesoes psoriaseformes clássicas, mas com tendência familiar, início precoce e tendem a formar placas grandes e convalescentes68,69. Nenhuma das duas tendem a ter acometimento articular e, quando em evoluçao crônica, formam grandes cicatrizes com atrofias cutâneas.
Doenças granulomatosas
Doenças granulomatosas (OMIM: Síndrome de Blau #186580)
A Síndrome de Blau está associada a mutaçoes no gene NOD2 70. A síndrome é caracterizada pela tríade de inflamaçao granulomatosa crônica de olhos, articulaçoes e pele. Artrite, geralmente simétrica, é sempre observada, sendo que a poliartrite é a mais prevalente. Uveíte é comum, afetando ambos os olhos. Também podem ocorrer: catarata, glaucoma e perda visual71,72. O exantema é de coloraçao marrom e ictiosiforme, que afeta face e tronco e pode descamar. Com menor frequência, ocorre febre, hepatoesplenomegalia e neuropatia. É comum haver aumento sérico de imunoglobulinas e PCR. O principal diagnóstico diferencial é a sarcoidose73,74.
Defeitos de MONARCH1 (OMIM: Síndrome Autoinflamatória relacionada ao Frio tipo 2 #609648)
Os defeitos deste grupo estao ligados a alteraçoes no gene NLRP12 75. Podem se manifestar, mais comumente, através de urticária induzida pelo frio, rash malar e úlceras em cavidade oral. Sintomas gerais como febre, mialgia, cefaleia e fadiga podem preceder as crises. Estao relacionados à perda auditiva neurossensorial. Sintomas menos comuns sao dor abdominal, lifonodomegalia e artralgia. Laboratorialmente, encontra-se aumento de PCR e outras proteínas de fase aguda. Ainda nao foi descrito amiloidose76,77.
Síndromes autoinflamatórias relacionadas a defeitos no proteossoma (PRASS - Proteassoma-Associated Autoinflammatory Syndromes)
Defeitos do proteossoma (OMIM: Síndrome da Autoinflamaçao, Lipodistrofia e Dermatoses #256040)
Sao um grupo espectral de doenças relacionado a mutaçoes em múltiplos genes como: PSMB8, PSMB4, PSMB9, PSMA3 e POMP. As doenças pertencentes a este grupo sao: Síndrome de Nakajo-Nishimura, Síndrome JMP (Joint Contractures, Muscular Atrophy, Mycrocitic Anemia and Panniculitis-Induced Lypodistrophy) e Síndrome CANDLE (Chronic Atypical Neutrophilic Dermatosis with Lypodistrophy and Elevated Temperature). As manifestaçoes clínicas entre elas sao variadas, mas com um ponto em comum a todas, o envolvimento cutâneo, denominado por dermatoses neutrofílicas. Elas sao clinicamente evidentes, com lesoes em placas associadas a lesoes purpúricas, eritema palpebral, lipodistrofia em face e também articulaçoes. Alguns pacientes têm febre alta constante. Na infância, levam ao atraso no crescimento e desenvolvimento. Podem causar meningite asséptica, episclerite, conjuntivite e ceratite, a qual pode evoluir para cegueira. Quando acometem o sistema de conduçao cardíaca podem levar a miocardiopatia dilatada e arritmias cardíacas. Otite, sinusite, diarreia, hepatomegalia, linfonodomegalia e aumento de enzimas hepáticas sao menos frequentes. Atrofia muscular, miosite e fadiga podem estar presentes. Laboratorialmente observamos, principalmente, anemia e plaquetose, além de aumento de provas inflamatórias78-80.
Outras doenças sem síndromes definidas
Doenças de ativaçao macrofágica (OMIM #267700)
Podem estar relacionadas ao HLA-DRB1 e mutaçoes nos seguintes genes: PRF1, STX11, STXBP2, MUNC13-4, RAB27A, SH2D1A e BIRC4. Podese encontrar clinicamente uma erupçao cutânea morbiliforme rosa-salmao típica associada à febre, prurido e mialgia. Neurologicamente, evidencia-se meningismo e sinais de hipertensao intracraniana que podem levar ao coma. Pleurite associada a derrame pleural, pericardite e miocardite podem estar presentes. Há predisposiçao a infecçoes respiratórias. Hepatoesplenomegalia, linfonofomegalia, artrite e artralgia também sao comuns. Amiloidose secundária é uma complicaçao possível. A alteraçao laboratorial mais comum é o aumento da ferritina81,82.
Doenças associadas ao PLCG2 (OMIM: Síndrome Autoinflamatória Relacionada ao Frio tipo 3 #614468 / APLAID: Autoinflammation, Antibody Deficiency and Immune Dysregulation#614878)
Sao doenças associadas a mutaçoes no gene PLCG2 83. A apresentaçao cutânea se dá através de placas exantemáticas e vesicopustulares que pioram à exposiçao solar (APLAID), ou urticária induzida pelo frio e angioedema (FCAS tipo 3). As manifestaçoes oculares compreendem o glaucoma, a erosao da córnea e a catarata. No aparelho respiratório observamos asma, sinusites recorrentes e pneumonia intersticial com bronquiolite, e no trato gastrointestinal, diarreia e colite ulcerativa. Alteraçoes laboratoriais encontradas: aumento da IgE e diminuiçao de IgA e IgM séricas, além de baixos níveis de células NK84,85.
Doenças relacionadas ao SLC29a3 (OMIM: Síndrome da Histiocitose com Linfonodomegalia #602782)
É uma doença recentemente descoberta e decorre de mutaçoes no gene SLC29a3. Todos pacientes apresentam linfonodomegalia maciça (por vezes generalizada) associada ao achado de histiocitose na biópsia (coloraçao por CD1a na imunohistoquímica). Outros achados que podem estar presentes sao: hiperpigmentaçao cutânea, hipertricose, características faciais dismórficas e atraso neuropsicomotor. As manifestaçoes oculares se dao por edema e eritema palpebral (cuja biopsia é de linfohistiocitose), uveíte e glaucoma. Perda auditiva neurossensorial é um achado frequente. Pericardite e defeitos septais e valvares cardíacos podem ocorrer. Ainda sao encontrados com certa frequência: diarreia, hepatoesplenomegalia, hipogonadismo, Diabetes mellitus, baixa estatura, artralgia e tórax escavado. Evidencia-se aumento de IgG durante as crises e alguns pacientes tem elevaçao de provas inflamatórias86-88.
Defeitos de ADA2 (OMIM: Deficiência de Adenosina Deaminase 2 - DADA2 #615688)
Trata-se de um grupo de doenças relacionadas à mutaçoes no gene CECR1. Clinicamente sao definidas por vasculopatia de início na infância, mas casos em adultos já foram relatados. A biópsia da lesao vasculítica sempre define PAN (poliarterite nodosa) e frequentemente há associaçao com eventos isquêmicos ou hemorrágicos cerebrais. Pode haver febre recorrente associada e é frequente o achado de livedo reticular. Hepatoesplenomegalia é comum. Laboratorialmente observamos pancitopenia e hipogamaglobulinemia89.
CONCLUSAO
As doenças autoinflamatórias sao doenças raras, de manifestaçoes clínicas particulares e típicas, que pertencem a prática clínica dos imunologistas e alergistas, sendo um diagnóstico diferencial importante a ser considerado. Nota-se que o conceito "autoinflamaçao" é muito mais amplo do que uma classe de doenças e deve ser aplicado em todas as áreas da medicina.
O Serviço de Imunologia Clínica e Alergia do Hospital das Clínicas da Universidade de Sao Paulo, de forma pioneira nesta especialidade, recentemente iniciou um ambulatório didático/científico voltado para o estudo e cuidado destas doenças, o que permitirá a identificaçao e abordagem adequada dos pacientes, além da descricao de novas mutaçoes. Como sao doenças de diagnóstico difícil, é necessária abordagem multidisciplinar e com base na pesquisa de traduçao (from bench to bed side and back again).
REFERENCIAS
1. Doria A, Dayer JM, Punzi L. Autoinflammatory diseases: How to put the fire inside the body out? Autoimmun Rev. 2012;12(1):1-4.
2. Galeazzi M, Gasbarrini G, Ghirardello A, Grandemange S, Hoffman HM, Manna R, et al. Autoinflammatory syndromes. Clin Exp Rheumatol. 2006;24(1 Suppl 40):S79-85.
3. Quintero-Ronderos P, Montoya-Ortiz G. Epigenetics and Autoimmune Diseases. Autoimmune Diseases. 2012;2012:593720.
4. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol. 2009;27:621-68.
5. Stojanov S, Kastner DL. Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol. 2005;17(5):586-99.
6. Aksentijevich I, Centola M, Deng Z, Sood R, Balow JEJ, Wood G, et al. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90(4):797-807.
7. Padeh S. Periodic fever syndromes. Pediatr Clin North Am. 2005;52:577-609.
8. Alghamdi M. Familial Mediterranean fever, review of the literature. Clin Rheumatol. 2017;36(8):1707-13.
9. Ozen S, Hoffman HM, Frenkel J, Kastner D. Familial Mediterranean fever (FMF) and beyond: a new horizon. Fourth International Congress on the Systemic Autoinflammatory Diseases held in Bethesda, USA, 6-10 November 2005. Ann Rheum Dis. 2006;65:961-4.
10. Galeazzi M, Gasbarrini G, Ghirardello A, Grandemange S, Hoffman HM, Manna R, et al. Autoinflammatory syndromes. Clin Exp Rheumatol. 2006;24(1 Suppl 40):S79-85.
11. Kiss MH, Magalhaes CS. Autoinflammatory diseases: mimics of autoimmunity or part of its spectrum? Case presentation. J Clin Immunol. 2008;28(Suppl 1):S84-9.
12. Medzhitov R, Janeway C Jr. Innate immunity. N Engl J Med. 2000;343:338-44.
13. Janeway CA, Medzhitov R. Innate immunity recognition. Annu Rev Immunol. 2002;20:197-216.
14. Areschoug T, Gordon S 2009. Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell Microbiol. 2009;11(8):1160-9.
15. Bakir HY, Tomiyama C, Abo T 2011. Cytokine profile of murine malaria: stage-related production of inflammatory and anti-inflammatory cytokines. Biomed Res. 2011;32(3):203-8.
16. Agnello D, Lankford CSR, Bream J, Morinobu A, Gadina M, Shea JJO, Frucht DM. Cytokines and transcription factors that regulate T helper cell differentiation: new players and new insights. J Clin Immunol. 2003;23(3):147-61.
17. Kobayashi K, Inohara N, Hernandez LD, Galán JE, Núñez G, Janeway CA, et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature. 2002;416:194-9.
18. Carpenter S, Ricci EP, Mercier BC, et al. Post-transcriptional regulation of gene expression in innate immunity. Nat Rev Immunol. 2014;14:361-76.
19. Barrington R, Zhang M, Fischer M, Carroll MC. The role of complement in inflammation and adaptive immunity. Immunol Rev. 2001;180:5-15.
20. O'Neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol. 2011;11:163-75.
21. Paro S, Imler J-L, Meignin C. Sensing viral RNAs by Dicer/RIG-I like ATPases across species. Current Opinion in Immunology. 2015;32:106-13.
22. Germain RN. MHC-dependent antigen processing and peptide presentation: providing ligands for T lymphocyte activation. Cell. 1994;76:287-99.
23. Kim YK, Shin J-S, Nahm MH. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Medical Journal. 2016;57(1):5-14.
24. Leonard CA, Schoborg RV, Borel N. Damage/Danger Associated Molecular Patterns (DAMPs) Modulate Chlamydia pecorum and C. trachomatis Serovar E Inclusion Development In Vitro. Kaltenboeck B, ed. PLoS ONE. 2015;10(8):e0134943.
25. Santoni G, Cardinali C, Morelli MB, Santoni M, Nabissi M, Amantini C. Danger- and pathogen-associated molecular patterns recognition by pattern-recognition receptors and ion channels of the transient receptor potential family triggers the inflammasome activation in immune cells and sensory neurons. Journal of Neuroinflammation. 2015;12:21.
26. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10(3):241-7.
27. Wang H, Liu S, Wang Y, Chang B, Wang B. Nod-like receptor protein 3 inflammasome activation by Escherichia coli RNA induces transforming growth factor beta 1 secretion in hepatic stellate cells. Bosn J Basic Med Sci. 2016;16(2):126-31.
28. Franchi L, Kanneganti TD, Dubyak GR, Núñez G. Differential requirement of P2X7 receptor and intracellular K+ for caspase-1 activation induced by intracellular and extracellular bacteria. J Biol Chem. 2007;282(26):18810-8.
29. Kanneganti TD, Ozören N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233-6.
30. Cassel SL, Joly S, Sutterwala FS. The NLRP3 inflammasome: a sensor of immune danger signals. Semin Immunol. 2009;21(4):194-8.
31. Abbas AK, Lichtman AH. Cellular and Molecular Immunology. 6th ed. Saunders; 2003.
32. Stych B, Dobrovolny D. Familial cold auto-inflammatory syndrome (FCAS)characterization of symptomatology and impact on patients' lives. Curr Med Res Opin. 2008;24(6):1577-82.
33. Gattorno M, Federici S, Pelagatti MA, Caorsi R, Brisca G, Malattia C, Martini A. Diagnosis and management of autoinflammatory diseases in childhood J Clin Immunol. 2008;28 (Suppl 1):S73-S8.
34. Johnstone RF, Dolen WK, Hoffman HM. A large kindred with familial cold autoinflammatory syndrome. Ann Allergy Asthma Immunol. 2003;90(2):233-7.
35. Wanderer AA, Hoffman HM. The spectrum of acquired and familial cold induced urticaria/urticaria like syndromes. Immunol Allergy Clin North Am. 2004;24(2):259-86.
36. Watts RA, Nicholls A, Scott DG. The arthropathy of the Muckle Wells syndrome. Br J Rheumatol. 1994;33(12):1184-7.
37. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin like protein causes familial cold autoinflammatory syndrome and Muckle Wells syndrome. Nat Genet. 2001;29(3):301-5.
38. Hentgen V, Despert V, Leprêtre AC, Cuisset L, Chevrant-Breton J, Jégo P, et al. Intrafamilial variable phenotypic expression of a CIAS1 mutation: from Muckle Wells to chronic infantile neurological cutaneous and articular syndrome. J Rheumatol. 2005;32(4):747-51.
39. Simon A, Asli B, Braun-Falco M, De Koning H, Fermand JP, Grattan C, et al. Schnitzler'ssyndrome: diagnosis, treatment, and follow-up. Allergy. 2013;68:562-8.
40. Lipsker D, Veran Y, Grunenberger F, Cribier B, Heid E, Grosshans E. The Schnitzler syndrome. Four new cases and review of the literature. Medicine (Baltimore). 2001;80:37-44.
41. Goupille P, Pizzuti P, Diot E, Jattiot F, Guilmot JL, Valat JP. Schnitzler's syndrome (urticaria and macroglobulinemia) dramatically improved with corticosteroids. Clin Exp Rheumatol. 1995;13:95-8.
42. Herráez Albendea MM, López Rodríguez M, López de la Guía A, Canales Albendea MA. Síndrome de Schnitzler. Reumatol Clin. 2013;9:383-5.
43. Nashan D, Sunderkotter C, Bonsmann G, Luger T, Goerdt S. Chronic urticaria, arthralgia, raised erythrocyte sedimentation rate and IgG paraproteinaemia: a variant of Schnitzler's syndrome? Br J Dermatol.1995;133:132-4.
44. Akimoto R, Moshida M, Matsuda R, Miyasaka K, Itoh M. Schnitzler's syndrome with IgG gammopathy. J Dermatol. 2002;29:735-8.
45. de Koning HD, Bodar EJ, van der Meer JW, Simon A; Schnitzler Syndrome Study Group. Schintzler's syndrome: beyond the case reports: review and follow-up of 94 patients with an emphasis on prognosis and treatment. Semin Arthritis Rheum. 2007;37:137-48.
46. Ben-Zvi I, Livneh A. Chronic inflammation in FMF: markers, risk factors, outcomes and therapy. Nat Rev Rheumatol. 2011;7(2):105-12.
47. Onen F. Familial Mediterranean fever. Rheumatol Int. 2006;26(6):489-96.
48. Ozkurede VU, Franchi L. Immunology in clinic review series; focus on autoinflammatory diseases: role of inflammasomes in autoinflammatory syndromes. Clinical and Experimental Immunology. 2012;167(3):382-90.
49. Jesus AA, Oliveira JB, Hilário Maria MOE, Terreri RA, Fujihira E, Watase M, et al . Síndromes autoinflamatórias hereditárias na faixa etária pediátrica. J Pediatr (Rio J). 2010;86(5):353-66.
50. Tunca M, Akar S, Onen F, Ozdogan H, Kasapcopur O, Yalcinkaya F, et al. Familial Mediterranean fever (FMF) in Turkey: results of a nation wide multicenter study. Medicine (Baltimore). 2005;84(1):1-11.
51. The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell. 1997;90(4):797-807.
52. Tricarico PM, Marcuzzi A, Pisciana E, Monasta L, Crovella S, Kleiner G. Mevalonate Kinase Deficiency and Neuroinflammation: Balance between Apoptosis and Pyroptosis. International Journal of Molecular Sciences. 2013;14(12):23274-88.
53. Henneman L, Schneiders MS, Turkenburg M, Waterham HR. Compromized geranylgeranylation of RhoA and Rac1 in mevalonate kinase deficiency. Journal of Inherited Metabolic Disease. 2010;33(5):625-32.
54. Haas D, Hoffmann GF. Mevalonate kinase deficiencies: from mevalonic aciduria to hyperimmunoglobulinemia D syndrome. Orphanet Journal of Rare Diseases. 2006;1:13.
55. Hinson DD, Ross RM, Krisans S, Shaw JL, Kozich V, Rolland MO, et al. Identification of a mutation cluster in mevalonate kinase deficiency, including a new mutation in a patient of Mennonite ancestry. Am J Hum Genet. 1999;65(2):327-35.
56. Mulders-Manders CM, Simon A. Hyper-IgD syndrome/mevalonate kinase deficiency: what is new? Seminars in Immunopathology. 2015;37(4):371-6.
57. Tricarico PM, Crovella S, Celsi F. Mevalonate pathway blockade, mitochondrial dysfunction and autophagy: a possible link. Int J Mol Sci. 2015;16(7):16067-84.
58. De Jesus AA, Goldbach-Mansky R. Monogenic Autoinflammatory diseases: concept and clinical manifestations. Clinical immunology (Orlando, Fla). 2013;147(3):155-74.
59. Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Annals of the Rheumatic Diseases. 2007;66(8):1113-5.
60. Arkwright PD, McDermott MF, Houten SM, Frenkel J, Waterham HR, Aganna E, et al. Hyper IgD syndrome (HIDS) associated with in vitro evidence of defective monocyte TNFRSF1A shedding and partial response to TNF receptor blockade with etanercept. Clin Exp Immunol. 2002;130(3):484-8.
61. Fujikawa K, Migita K, Shigemitsu Y, Umeda M, Nonaka F, Tamai M, et al. MEFV gene polymorphisms and TNFRSF1A mutation in patients with inflammatory myopathy with abundant macrophages. Clin Exp Immunol. 2014;178(2):224-8.
62. Pelagatti MA, Meini A, Caorsi R, Cattalini M, Federici S, Zulian F, et al. Long-Term Clinical Profile of Children With the Low-Penetrance R92Q Mutation of the TNFRSF1A Gene. Arthritis and Rheumatism. 2011;63(4):1141-50.
63. Greco E, Aita A, Galozzi P, et al. The novel S59P mutation in the TNFRSF1A gene identified in an adult onset TNF receptor associated periodic syndrome (TRAPS) constitutively activates NF-κB pathway. Arthritis Research & Therapy. 2015;17(1):93.
64. Jesus AA, Osman M, Silva CA, Kim PW, Pham TH, Gadina M, et al. A novel mutation of IL1RN in the deficiency of Interleukin-1 receptor antagonist Syndrome: Description of two unrelated cases from Brazil. Arthritis Rheum. 2011;63(12):4007-17.
65. Lukens JR, Gurung P, Vogel P, Johnson GR, Carter RA, McGoldrick DJ, et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature. 2014;516(7530):246-9.
66. Stern SM, Ferguson PJ. Autoinflammatory Bone Diseases. Rheumatic diseases clinics of North America. 2013;39(4):735-49.
67. Wang D, Höing S, Patterson HC, Ahmad UM, Rathinam VA, Rajewsky K, et al. Inflammation in Mice Ectopically Expressing Human Pyogenic Arthritis, Pyoderma Gangrenosum, and Acne (PAPA) Syndrome-associated PSTPIP1 A230T Mutant Proteins. J Biol Chem. 2013;288(7):4594-601.
68. Yu JW, Fernandes-Alnemri T, Datta P, Wu J, Juliana C, Solorzano L, et al. Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol Cell. 2007;28(2):214-27.
69. Grosse J, Chitu V, Marquardt A, Hanke P, Schmittwolf C, Zeitlmann L, et al. Mutation of mouse Mayp/Pstpip2 causes a macrophage autoinflammatory disease. Blood. 2006;107(8):3350-8.
70. Wouters CH, Maes A, Foley KP, Bertin J, Rose CD. Blau Syndrome, the prototypic auto-inflammatory granulomatous disease. Pediatr Rheumatol Online J. 2014;12:33.
71. Dugan J, Griffiths E, Snow P, Rosenzweig H, Lee E, Brown B, et al. A Blau syndrome-associated Nod2 mutation alters expression of full length NOD2 and limits responses to muramyl dipeptide in knock-in mice. J Immunol. 2015;194(1):349-57.
72. Parkhouse R, Boyle JP, Monie TP. Blau syndrome polymorphisms in NOD2 identify nucleotide hydrolysis and helical domain 1 as signalling regulators. Febs Letters. 2014;588(18):3382-9.
73. Martin TM, Zhang Z, Kurz P, Rosé CD, Chen H, Lu H, et al. The NOD2 defect in Blau syndrome does not result in excess interleukin 1 activity. Arthritis Rheum. 2009;60(2):611-8.
74. Caso F, Galozzi P, Costa L, Sfriso P, Cantarini L, Punzi L. Autoinflammatory granulomatous diseases: from Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn's disease. RMD Open. 2015;1(1):e000097.
75. Franchi L, Warner N, Viani K, Nuñez G. Function of nod-like receptors in microbial recognition and host defense. Immunological reviews. 2009;227(1):106-28.
76. Sutterwala FS, Flavell RA. NLRC4/IPAF: a CARD carrying member of the NLR family. Clinical immunology (Orlando, Fla). 2009;130(1):2-6.
77. Catalytic Receptors. Br J Pharmacol. 2011;164(Suppl 1):S189-S212.
78. Sjakste T, Paramonova N, Wu LS, Zemeckiene Z, Sitkauskiene B, Sakalauskas R, et al. PSMA6 (rs2277460, rs1048990), PSMC6 (rs2295826, rs2295827) and PSMA3 (rs2348071) genetic diversity in Latvians, Lithuanians and Taiwanese. Meta Gene. 2014;2:283-98.
79. Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011;121(10):4150-60.
80. Gomes AV. Genetics of Proteasome Diseases. Scientifica. 2013;2013:637629.
81. Belcastro E, Franzini M, Cianchetti S, Lorenzini E, Masotti S, Fierabracci V, et al. Monocytes/macrophages activation contributes to b-gamma-glutamyltransferase accumulation inside atherosclerotic plaques. J Trans Med. 2015;13:325.
82. Loyo E, Jara LJ, López PD, Puig AC. Autoimmunity in connection with a metal implant: a case of autoimmune/autoinflammatory syndrome induced by adjuvants. Auto-Immunity Highlights. 2013;4(1):33-8.
83. Chae JJ, Park YH, Park C, Hwang IY, Hoffmann P, Kehrl JH, et al. Connecting Two Pathways through Ca2+ Signaling: NLRP3 Inflammasome Activation Induced by a Hypermorphic PLCG2 Mutation. Arthritis Rheumatol. 2015;67(2):563-7.
84. Zhou Q, Lee GS, Brady J, Datta S, Katan M, Sheikh A, et al. A Hypermorphic Missense Mutation in PLCG2, Encoding Phospholipase Cγ2, Causes a Dominantly Inherited Autoinflammatory Disease with Immunodeficiency. Am J Hum Genet. 2012;91(4):713-20.
85. Ombrello MJ, Remmers EF, Sun G, Freeman AF, Datta S, Torabi-Parizi P, et al. Cold Urticaria, Immunodeficiency, and Autoimmunity Related to PLCG2 Deletions. N Engl J Med. 2012 Jan 26;366(4):330-8.
86. Cliffe ST, Kramer JM, Hussain K, Robben JH, de Jong EK, de Brouwer AP, et al. SLC29A3 gene is mutated in pigmented hypertrichosis with insulin-dependent diabetes mellitus syndrome and interacts with the insulin signaling pathway. Hum Mol Genet. 2009;18(12):2257-65.
87. Morgan NV, Morris MR, Cangul H, Gleeson D, Straatman-Iwanowska A, Davies N, et al. Mutations in SLC29A3, encoding an equilibrative nucleoside transporter ENT3, cause a familial histiocytosis syndrome (Faisalabad histiocytosis) and familial Rosai-Dorfman disease. PLoS Genet. 2010;6(2):e1000833.
88. Spiegel R, Cliffe ST, Buckley MF, Crow YJ, Urquhart J, Horovitz Y, et al. Expanding the clinical spectrum of SLC29A3 gene defects. Eur J Med Genet. 2010;53(5):309-13.
89. Tadashi A, Noriko O, Koji Y, Nobuaki K, Hideaki K, Shoichiro T, et al. T-cell lines from 2 patients with adenosine deaminase (ADA) deficiency showed the restoration of ADA activity resulted from the reversion of an inherited mutation. Blood May. 2001;97(9):2896-29.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888