Número Atual: Fevereiro- 2015 - Volume 3 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

RELATO DE CASO
Deficiência do componente C5 do complemento associada a meningites meningocócicas
Complement component C5 deficiency associated with meningococcal meningitis
Wilma C. Neves Forte1; Tainá Mosca2; Vitor A. P. Mazon3; Elias J. E. Ghosn3; Raphael J. P. Fins3; Carlos A. Longui1; Maria da Conceiçao S. de Menezes4
1. MD, PhD. Faculdade de Ciências Médicas da Santa Casa de Sao Paulo
2. PhD. Faculdade de Ciências Médicas da Santa Casa de Sao Paulo
3. MD. Faculdade de Ciências Médicas da Santa Casa de Sao Paulo
4. MD, MSc. Irmandade da Santa Casa de Misericórdia de Sao Paulo
Endereço para correspondência:
Wilma Carvalho Neves Forte
E-mail: wilmanevesforte@yahoo.com.br
Submetido em: 18/6/2015.
Aceito em 22/11/2015.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
O presente estudo teve como objetivo relatar o caso de paciente com infecçoes meningocócicas graves, avaliar seu sistema complemento e o de 22 membros de sua família. Foram quantificados o complemento total (CH50) e os componentes C3, C4 e C5 do complemento. Foi observado que a paciente e quatro familiares apresentavam deficiência do componente C5: dois haviam apresentado meningites meningocócicas graves e um apresentou a infecçao logo após o diagnóstico da deficiência. Concluímos que a paciente com história de infecçoes meningocócicas graves apresentou deficiência do componente C5 do complemento, assim como quatro de seus familiares. Nossos resultados indicam que a avaliaçao do sistema complemento em portadores de infecçoes meningocócicas e em familiares próximos deva ser realizada para identificar pacientes de risco.
Descritores: Imunodeficiência, complemento C5, meningite meningocócica, Neisseria.
INTRODUÇAO
A deficiência do complemento faz parte das imunodeficiências primárias (IDPs)1, e caracteriza-se por diminuiçao de qualquer componente da cascata do sistema complemento. A deficiência dos componentes iniciais (C1, C4 e C2) está mais relacionada à presença de doenças autoimunes em baixa idade2,3, possivelmente pela localizaçao dos genes responsáveis pela codificaçao destes componentes. A deficiência dos componentes terminais (C5, C6, C7, C8 e C9) tem sido associada a infecçoes por bactérias do gênero Neisseria, em especial N. meningitidis2,3. A deficiência de C3 pode levar tanto a doenças autoimunes, como a infecçoes meningocócicas.
As deficiências de complemento estao estimadas em 1 a 10% das IDPs4. A associaçao entre infecçao meningocócica e deficiência dos componentes terminais do complemento é alta, podendo chegar a 66% dos casos5. Acredita-se que 39% dos pacientes com deficiência dos componentes terminais do complemento apresentem pelo menos um episódio de infecçao meningocócica durante a vida, sendo mais descritas as deficiências de C6 a C9. As infecçoes meningocócicas de tais pacientes muitas vezes sao dadas por sorotipos nao usuais de Neisseria meningitidis6. Estudo de prevalência da doença meningocócica por sorotipos nao usuais de N. meningitidis mostrou que 33% dos pacientes apresentavam alguma deficiência do sistema complemento7.
Até o momento estao descritos na literatura menos de 60 casos de deficiência de C5. No Brasil há um relato de três membros de uma família com deficiência de C58 e de um adulto com três episódios de meningite meningocócica associados à deficiência de C89. É provável que a deficiência do complemento seja subdiagnosticada em nosso meio, tendo em vista a alta prevalência de meningite meningocócica e os poucos casos descritos dessa IDP. O diagnóstico da deficiência de complemento permite conduta terapêutica adequada e melhor prognóstico dos portadores.
O presente estudo teve como objetivo quantificar o sistema complemento de paciente com infecçao meningocócica grave e de seus familiares, procurando possíveis deficiências do complemento.
DESCRIÇAO DO CASO
Menina de nove anos, parda, procedente de Poá, Sao Paulo, Brasil, com história de duas meningites meningocócicas graves, segundo a mae. O primeiro quadro foi aos dois anos, com 33 dias de internaçao (três em UTI, necessitando de intubaçao). Durante tal episódio evoluiu com necrose do hálux, sendo necessária amputaçao. O segundo quadro ocorreu aos nove anos, permanecendo internada por 27 dias (6 em UTI, com intubaçao). Nesse episódio apresentou necrose em vários locais em infecçao de pele, que evoluíram para queloides. Os pais sao primos em primeiro grau. Havia história de meningite em prima materna, além de tio e primo paternos.
Os exames imunológicos da paciente mostraram resultados em relaçao à faixa etária: imunoglobulinas séricas normais, titulaçoes positivas de anticorpos contra vários sorotipos de S. pneumoniae, complemento total (CH50) 110 U/mL (valor normal 130 a 330 U/mL), C3 108 mg/dL (valor normal 88 a 201 mg/dL), C4 23 mg/dL (valor normal 16 a 47 mg/dL), C5 sem formaçao de halo por imunodifusao radial (Binding SiteR - Birmingham, Reino Unido) e C5 0,9 mg/L (valor normal 45 a 190 mg/L) por ELISA.
Foi feita investigaçao imunológica do irmao: CH50 diminuído, C5 sem formaçao de halo por imunodifusao radial e 1,0 mg/L por ELISA. Logo após a investigaçao imunológica, o irmao apresentou meningite meningocócica, recebendo antibiótico e plasma fresco congelado, tendo boa evoluçao e alta sem sequelas.
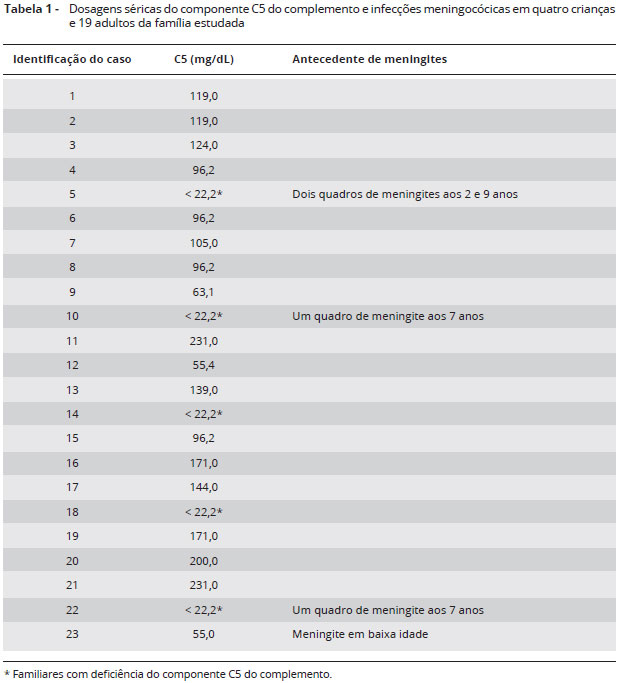
Além da paciente e do irmao, foram selecionados 21 familiares: pais, tios e primos. Entre todas as amostras analisadas (Tabela 1), cinco apresentaram diminuiçao de CH50 e C5 abaixo de 22,2 mg/L por imunodifusao radial: paciente, irmao, pai, prima e tio paternos. Foi pesquisada deleçao no exon 30 nos familiares afetados, nao sendo observada tal deleçao.
A paciente e seus familiares nao haviam recebido vacina antimeningocócica até o presente estudo. A partir de entao, todos os membros da família foram orientados à vacinaçao.
O presente estudo foi aprovado pelo Comitê de Ética da Instituiçao, sob o número 20710, com o consentimento assinado dos indivíduos estudados e/ou de seus responsáveis.
DISCUSSAO
A observaçao de deficiência do componente C5 na paciente índice foi coerente com os dados clínicos apresentados de história de duas internaçoes por infecçoes meningocócicas graves. Os exames iniciais da paciente foram dirigidos para a avaliaçao do complemento, uma vez que este sistema é necessário para a defesa contra Neisseria meningitidis3,4. Foram feitas as dosagens de imunoglobulinas séricas, pois as IDPs mais frequentes sao por deficiências predominantemente de anticorpos.
O sistema complemento é um conjunto de pelo menos 30 proteínas plasmáticas e de superfície celular. Sua principal atividade biológica é a lise de células infectadas ou de micro-organismos.
Estao descritas três vias de ativaçao do sistema complemento: clássica, alternativa e das lectinas. A via clássica é ativada por imunoglobulinas (IgM, IgG1 ou IgG3). A uniao do antígeno ao terceiro domínio da imunoglobulina específica determina a ativaçao de C1q, seguindo-se C1r e C1s. O complexo C1qrs cliva C4 e o componente C4b continua a cascata, com ativaçao de C2. O complexo C1qrs4b2a, atuando como uma C3 convertase, cliva o componente C3 nas fraçoes C3a e C3b. O novo complexo C4b2a3b é uma C5 convertase, capaz de cindir C5 em C5a e C5b. O componente C5b permanece na cascata, com ativaçao sequencial de C6, C7, C8 e C910. Os componentes terminais C5b6789 formam o complexo de ataque à membrana (MAC). A ativaçao desses componentes terminais resulta em alteraçao funcional de fosfolipídios da membrana. Há formaçao de um canal que facilita a entrada de água, havendo intumescimento da célula ou do micro-organismo, e lise. Os componentes C3a e C5a, formados durante a ativaçao do sistema, sao anafilatoxinas e fatores quimiotáticos para fagócitos; C3b e C5b sao opsoninas, que ao revestirem certos micro-organismos, facilitam o processo de fagocitose. Na via alternativa, o componente C3 é ativado diretamente. Na via das lectinas, a lectina do organismo une-se à manose de patógenos, dando sequência ao sistema complemento de forma análoga à via clássica10. Em todas as vias o componente C3 é necessário para a ativaçao dos componentes finais, enquanto o componente C5 tem papel crucial na formaçao do MAC. Os componentes C5b6789 sao comuns às três vias de ativaçao.
O complemento total ou CH50 é medido em unidades hemolíticas, refletindo a habilidade do soro testado em determinar 50% de lise de uma suspensao padronizada de hemácias de carneiro recobertas por soro de coelho. A observaçao de CH50 diminuído, C3 e C4 normais, levou à hipótese de que algum dos componentes finais pudesse estar diminuído. Nas deficiências congênitas dos componentes C1 a C8, o valor de CH50 costuma ser zero ou próximo de zero, enquanto que na deficiência de C9, tal valor é de aproximadamente 30 a 50% do valor normal4. A dosagem de C5 mostrou a deficiência deste componente - sem formaçao de halo por imunodifusao radial3, confirmado por diminuiçao da dosagem por método ELISA8. O componente C5 é um heterodímero, com pontes dissulfídicas inter-cadeia, com peso molecular de 190 kDa e sua ausência impede a formaçao do MAC.
É necessário que o sistema complemento esteja em perfeita integridade e funcionalidade para que bactérias do gênero Neisseria sejam eliminadas, sendo a erradicaçao feita principalmente através dos componentes terminais C5b6789 (MAC)10. A Neisseria meningitidis, um diplococo encapsulado Gram-negativo, necessita ser revestida pelos componentes C3b e C5b para ser opsonizada, mas a principal defesa contra tal bactéria é dada por lise extracelular, através do MAC. Este complexo também é necessário para defesa contra Neisseria gonorrhoeae, podendo ocorrer gonorreia extragenital na deficiência de MAC. Além disso, sabe-se que o componente C5a liga-se ao receptor C5a-R que pertence à família das proteínas G, para ativaçao da proteína quinase C (PKC). As PKCs ativadas fosforilam a proteína p47-phox, iniciando a cascata do complexo phox nos fagolisossomos. Pesquisadores observaram que macrófagos C5-/- possuíam C5a-R íntegros e funcionantes, porém incapazes de fosforilar certas isoformas de PKC, levando a reduzida síntese de ROS; consequentemente, os macrófagos tornavam-se ineficazes em lisar as bactérias fagocitadas11.
Continuou-se a investigaçao com o único irmao da paciente, que apresentou exames com deficiência do componente C5. Os pais foram orientados a procurarem hospital, caso a criança tivesse infecçoes. Logo após o diagnóstico da deficiência, este irmao da paciente apresentou meningite meningocócica e pôde ser tratado de forma adequada para esta IDP: além de antibióticos, recebeu plasma fresco congelado, propiciando uma rápida melhora. O plasma, contendo componentes do complemento, é indicado em casos de deficiências dos componentes terminais, quando em quadros infecciosos que necessitem da defesa por complemento. O plasma administrado deve ser fresco e congelado, pois os componentes do complemento sao termolábeis.
A deficiência de C5 foi observada também em outros três membros da família, dentre os quais, uma prima que havia apresentado meningite meningocócica. O pai e o tio paterno, também com deficiência de C5, nao haviam apresentado infecçao meningocócica. É possível que pai e tio nao tenham manifestado a doença por terem pouco contato com seus filhos, morando em locais diferentes dos filhos. Ao contrário, um primo da paciente, sem deficiência de C5 mas com convívio muito próximo à paciente, também apresentou meningite meningocócica. Sabe-se que a transmissao da Neisseria é por contato íntimo, através de perdigotos provenientes de nasofaringe de doente ou de portador assintomático. É provável que a Neisseria meningitidis estivesse colonizando membros da família12 e que algum evento individual levou ao desencadeamento da doença. Durante este estudo, todos os familiares foram orientados quanto à vacinaçao antimeningocócica e reforço, como indicado em nosso meio, na tentativa de diminuir tanto a doença meningocócica como o estado de portador13.
A investigaçao genética foi baseada na mutaçao descrita nos três casos de deficiência de C5 do nosso meio: deleçao homozigota que corresponde precisamente ao exon 30, encontrada no sequenciamento do cDNA codificando C5. O sequenciamento da regiao correspondente do DNA genômico mostrou substituiçao de GAG4028 por GAA4028 na regiao 3' do exon, levando a um erro de splicing do exon 30, e resultando em síntese de proteína truncada seguida de degradaçao8. Após amplificaçao e sequenciamento utilizando primers externos à possível deleçao, concluímos que os cinco membros com deficiência de C5 da família relatada neste estudo nao apresentaram tal deleçao. Entre quase 60 casos descritos na literatura atual com deficiência de C5, a base molecular foi descrita em poucos casos, encontrando-se diferentes mutaçoes8,14,15.
A deficiência de C5 está mais descrita em países desenvolvidos, com baixa prevalência da infecçao meningocócica. O possível subdiagnóstico desta IDP em nosso meio foi o motivo de descrever a família que estudamos. Este estudo permitiu conduta adequada, em especial no irmao da paciente, com antibioticoterapia associada ao plasma fresco frente à infecçao por N. meningitidis.
Concluímos que a paciente com história de infecçoes meningocócicas graves apresentou deficiência do componente C5, assim como quatro de seus familiares apresentavam tal deficiência. Nosso resultados indicam ser necessária a avaliaçao do sistema complemento em portadores de infecçoes meningocócicas e em familiares próximos para identificar pacientes de risco.
REFERENCIAS
1. Notarangelo LD. Primary Immunodeficiencies. J Allergy Clin Immunol. 2010;125:S182-94.
2. Brazilian Group for immunodeficiency. [site na internet]. Disponível: http://imunopediatria.org.br/. Acessado: 30 de outubro de 2014.
3. The European Society for Immunodeficiencies registry. [site na internet]. Disponível: http://esid.org/Working-Parties/Registry/New-ESID-Registry. Acessado: 30 de outubro de 2014.
4. Grumach AS, Kirschfink M. Are complement deficiencies really rare? Overview on prevalence, clinical importance and modern diagnostic approach. Mol Immunol. 2014;61:110-7.
5. Lima Filho AB, Silva AMR, Moura P, Sarinho ESC. Meningite meningocócica, lecitina ligadora de manose, deficiência do complemento. Rev bras alerg imunopatol. 2011;34:3-6.
6. Iturry-Yamamoto GR, Portinho CP. Sistema complemento: ativaçao, regulaçao e deficiências congênitas e adquiridas. Rev Assoc Med Bras. 2001;47:41-51.
7. Figueroa JE, Densen P. Infectious diseases associated with complement deficiencies. Clin Microbiol Rev. 1991;4:359-95.
8. Aguilar-Ramirez P. Reis ES, Florido MPC, Barbosa AS, Farah CS, Costa-Carvalho BT, et al. Skipping of exon 30 in C5 gene results in complete human C5 deficiency and demonstrates the importance of C5d and CUB domains for stability. Mol Immunol. 2009;46:2116-23.
9. Rosa DD, Pasqualotto AC, Quadros M, Prezzi SH. Deficiency of the eighth component of complement associated with recurrent meningococcal meningitis - case report and literature review. Braz J Infect Dis. 2004;8:328-30.
10. Forte WCN. Imunologia do básico ao aplicado. 3ª ed. Sao Paulo: Editora Atheneu; 2015. 339p.
11. Daniel DS, Dai G, Singh CR, Lindsey DR, Smith AK, Dhandayuthapani S, et al. The reduced bactericidal function of complement C5-deficient murine macrophages is associated with defects in the synthesis and delivery of reactive oxygen radicals to mycobacterial phagosomes. J Immunol. 2006; 177(7):4688-98.
12. Sáfadi MA, Bettinger JA, Maturana GM, Enwere G, Borrow R, Global Meniingococcal Initiative. Evolving meningococcal immunization strategies. Expert Rev Vaccines. 2015;14:505-17.
13. Sáfadi MAP. Perspectivas na prevençao da doença meningocócica no Brasil. Rev Imunizaçoes (Soc Bras Imunizaçoes). 2014;7:8-11.
14. Schejbel L, Fadnes D, Permin H, Lappegard KT, Garred P, Mollnes TE. Primary complement C5 deficiencies - molecular characterization and clinical review of two families. Immunobiology. 2013;218(10):1304-10.
15. Delgado-Servino E, Fontán G, Lopez-Trascasa M. C5 complement deficiency in a Spanish family. Molecular characterization of the double mutation responsible for the defect. Mol Immunol. 2005;42:105-11.
Este trabalho recebeu apoio bolsa PIBIC para Vitor Augusto Petrili Manzon e apoio do Fundo de Amparo à Pesquisa para a realizaçao dos exames imunológicos para dosagens de C5 e dos exames genéticos.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888