Número Atual: Março-Abril 2014 - Volume 2 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo Original
Imunodeficiência combinada grave: uma revisão da literatura
Severe combined immunodeficiency: review of the literature
Juliana Cantagalli Pfisterer1; Sabrina Vargas Martini2; Paolo Ruggero Errante3; Josias Brito Frazao3
1. BSc. Departamento de Ciências Biomédicas, Centro Universitário Plínio Leite, Niterói, RJ
2. PhD. Instituto de Biofísica Chagas Filho, Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ
3. PhD. Instituto de Ciências Biomédicas, Universidade de Sao Paulo (ICB-USP), Sao Paulo, SP
Endereço para correspondência:
Josias Brito Frazao
E-mail: britoj@usp.br
Submetido em 22/06/2014
Aceito em 09/05/2015.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
Agências financiadoras: CAPES/CNPq, FAPESP.
RESUMO
A imunodeficiência combinada grave (SCID) é uma condiçao clínica caracterizada por marcante comprometimento da resposta imune envolvendo linfócitos T e/ou B e/ou células NK, que conduz a aumento da susceptibilidade a infecçoes e alta taxa de mortalidade em crianças acometidas. Dificuldades na interpretaçao dos sintomas clínicos e na identificaçao de mutaçoes genéticas, devido à ampla variedade fenotípica e genotípica da doença, representam obstáculos para o diagnóstico. Por outro lado, o tratamento é realizado de forma independente da identificaçao de mutaçao genética. O objetivo do presente trabalho foi revisar aspectos fisiopatológicos, métodos diagnósticos e tratamentos utilizados em pacientes com SCID. A revisao foi realizada com base em levantamento bibliográfico de banco de dados indexados disponíveis na Internet incluindo LILACS, MEDLINE, PubMed, SciELO Brasil, periódicos CAPES e Cochrane, e foi conduzida com os seguintes critérios de inclusao: artigos científicos publicados nos idiomas português e inglês, dentro do período de 1963 a 2014 e que possuíam as palavras-chave "Imunodeficiência Combinada Grave", "SCID", "Leucopenia", "Diagnóstico", "Tratamento" e "Transplante de medula óssea". O levantamento bibliográfico revelou dificuldades no diagnóstico clínico, laboratorial e genético-molecular, e ressaltou a importância do diagnóstico precoce conduzindo ao tratamento adequado. O diagnóstico precoce da SCID tem papel crucial na melhora da qualidade de vida e na sobrevida dos pacientes, além de favorecer intervençoes terapêuticas que previnem o surgimento de infecçoes e complicaçoes clínicas subsequentes.
Descritores: Imunodeficiência combinada grave, SCID, leucopenia, diagnóstico, tratamento, transplante de medula óssea.
INTRODUÇÃO
A imunodeficiência combinada grave (do inglês, Severe Combined Immunodeficiency - SCID) é uma imunodeficiência primária (IDP) com diversas causas genéticas que acarretam deficiência grave das funções desempenhadas por linfócitos T e/ou B. Em algumas formas da doença há também o comprometimento da função de células Natural Killers (NK)1. A classificação da SCID é baseada na presença ou ausência desses três tipos celulares, de acordo com as quais os pacientes são agrupados2,3.
Imunodeficiência combinada grave foi relatada pela primeira vez em 1950 por pesquisadores suíços4. O relato compreendia crianças que apresentavam uma grave linfopenia e evoluíam para óbito devido a infecções secundárias antes mesmo de seu primeiro ou segundo ano de vida. Com o passar do tempo, os diferentes padrões de herança encontrados na doença possibilitaram indicar uma variedade de causas genéticas para seu desenvolvimento. Atualmente, é sabido que as diversas formas de SCID são herdadas de forma autossômica recessiva ou de forma ligada ao cromossomo X (também conhecida por SCID-X) e que essas mutações podem ser classificadas em quatro fenótipos distintos5, descritos abaixo:
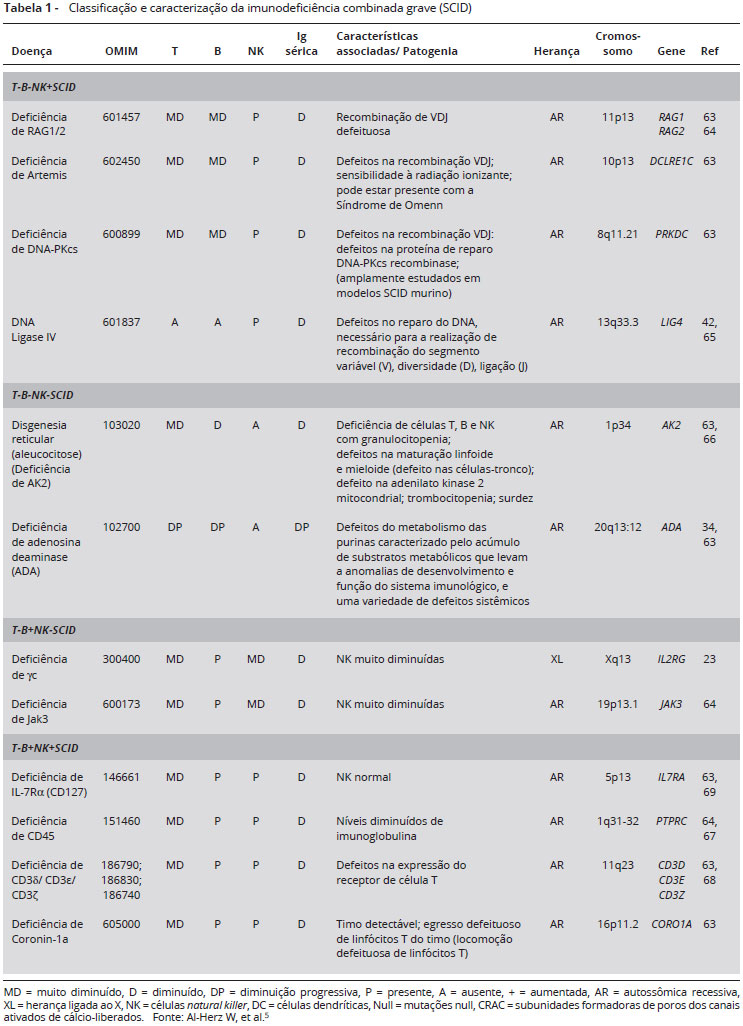
Ausência de linfócitos T e B (SCID T-B-NK+): encontram-se agrupadas aqui as deficiências de RAG 1 e 2 (RAG1/2), de Artemis (DCLRE1C), da subunidade catalítica da proteína-quinase DNA-dependente (DNA-PKcs) (PRKDC), de DNA Ligase IV (LIG4) e de Cernunnos/fator XRCC-4 (XLF).
Ausência de linfócitos T, B e NK (SCID T-B-NK-): neste grupo estão incluídas a disgenesia reticular com deficiência da adenilato quinase 2 (AK2) e a deficiência da adenosina deaminase (ADA).
Ausência de linfócitos T e células NK (SCID T-B+NK-): os defeitos genéticos que provocam ausência de linfócitos T e células NK incluem mutações na cadeia gama comum (γc) (IL2RG) e na JAK3 (JAK3).
Ausência de linfócitos T (SCID T-B+NK+): dentre as formas de SCID que estão agrupadas dentro deste fenótipo estão incluídas as deficiências da cadeia alfa do receptor de IL-7 (IL-7Rα - CD127), do CD45 (também chamado de proteína de tirosina fosfatase receptor do tipo c - PTPRC), do complexo CD3 (CD3δ, CD3ε, CD3ζ) e da proteína reguladora de actina (coronin 1a - CORO1A).
A Figura 1 ilustra a classificação das formas de SCID baseada na presença de linfócitos T e B, e células NK e sua caracterização.
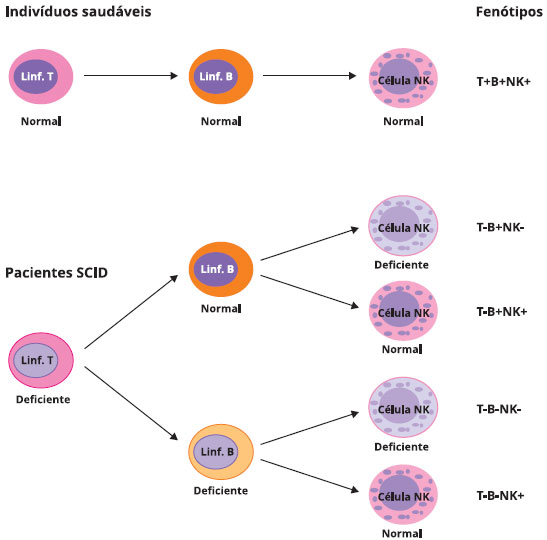
Figura 1 - A classificação dos fenótipos de imunodeficiência combinada grave (SCID) é realizada de acordo com o componente imunológico que se mostra alterado. Pacientes com SCID são classificados de acordo com a presença ou ausência de linfócitos T, linfócitos B e células NK, formando quatro fenótipos principais: SCID T-B+NK-, SCID T-B+NK+, SCID T-B-NK-, e SCID T-B-NK+. Indivíduos saudáveis apresentam o fenótipo T+B+NK+, representando a presença de linfócitos T, B e NK em níveis normais
Apesar dos avanços no entendimento desta doença, o diagnóstico precoce da SCID ainda representa um desafio em saúde pública. Progressos realizados no diagnóstico e tratamento contribuem para o controle dos sintomas e aumento da expectativa de vida dos pacientes acometidos por esta enfermidade6.
Foi objetivo deste trabalho revisar os aspectos fisiopatológicos, métodos diagnósticos e tratamentos utilizados em pacientes com imunodeficiência combinada grave. A revisão foi realizada com base no levantamento bibliográfico de banco de dados indexados disponíveis na Internet incluindo LILACS, MEDLINE, PubMed, SciELO Brasil, periódicos CAPES e Cochrane, e conduzida com os seguintes critérios de inclusão: artigos científicos publicados nos idiomas português e inglês, dentro do período de 1963 a 2014. A busca baseou-se nas palavras-chave: "Imunodeficiência Combinada Grave", "SCID", "Leucopenia", "Diagnóstico", "Tratamento" e "Transplante de medula óssea". Após a identificação dos títulos e obtenção de resumos, avaliados pelo autor, os artigos considerados relevantes foram incluídos como fonte bibliográfica do manuscrito.
DIAGNÓSTICO DE IMUNODEFICIÊNCIA COMBINADA GRAVE (SCID)
O diagnóstico da SCID é frequentemente iniciado com base em uma história clínica completa, exame físico e exames laboratoriais solicitados para dar base à investigação genético-molecular7.
Diagnóstico clínico
A SCID é considerada uma emergência médica pediátrica com perfil clínico heterogêneo, sendo que a anamnese e a investigação da história familiar têm papel importante na suspeita clínica da doença1,8. Tendo em vista que a maioria dos casos de SCID apresenta padrões de herança autossômica recessiva ou ligada ao X, é importante que a investigação da história familiar seja focalizada em relatos de membros da família com suscetibilidade a infecções, manifestações autoimunes e desenvolvimento de tumores. Os casos de consanguinidade favorecem a doença. Presença de linfopenia, febres recorrentes, retardo de crescimento, diarreia crônica, infecções graves recorrentes por vírus respiratório sincicial, herpes simples, varicela zoster, influenza e parainfluenza, além de reações adversas a vacinas de patógenos atenuados (Bacillus Calmette-Guérin BCG, rotavírus ou varicela) são fatores adicionais para conduzir a investigação imunológica9.
Crianças com SCID são frequentemente consideradas saudáveis ao nascer, porém apresentam susceptibilidade a infecções, que podem evoluir para septicemia10. Sinais de alerta incluem candidíase oral, eritema cutâneo, diarreia, retardo do crescimento e pneumonia intersticial9. Manifestações otorrinolaringológicas incluem congestão nasal, úlceras orais, adenopatia cervical, otite média e mastoidite11. Infecção respiratória persistente com pneumonia intersticial por Pneumocystis jirovecii, Citomegalovírus (CMV) e Aspergillus é frequente. Além disso, nódulos cutâneos e lesões nodulares em diferentes órgãos podem estar presentes em pacientes com imunodeficiência combinada grave12.
Crianças que apresentam essa imunodeficiência podem evoluir para óbito em idade precoce - na ausência de terapia com menos de 12 meses de vida - em decorrência de infecções causadas por agentes vacinais, como rotavírus, varicela e BCG13. O uso de vacinas com agentes vivos é contraindicado em pacientes com SCID. Havendo confirmação da exposição a esse tipo de vacinação, deve-se iniciar o tratamento adequado a cada tipo de vacina, mesmo na ausência de manifestações clínicas. Portanto, o diagnóstico de SCID é frequentemente realizado após o surgimento de infecções graves por Candida albicans, Pneumocystis jirovecii, varicela, adenovírus, vírus respiratório sincicial, parainfluenza, CMV, vírus Epstein-Barr (EBV), e após a administração da vacina BCG14.
O exame clínico de um paciente com SCID com a forma clássica da doença revela hipoplasia de tecidos linfoides (linfonodos, amígdalas) e ausência de sombra do timo na radiografia de tórax. A avaliação histológica em crianças com SCID demonstra hipoplasia tímica sem distinção corticomedular, baixo número de corpúsculos de Hassall e perda progressiva da função de maturação dos linfócitos T15. Entretanto, o timo de pacientes com SCID possui a capacidade de realizar o desenvolvimento normal dos linfócitos T mediante o fornecimento de células-tronco hematopoiéticas capazes de se desenvolver na medula óssea e se transformarem em timócitos. Além do timo, outros tecidos linfoides são desprovidos de linfócitos, como a polpa branca do baço. Sendo assim, o exame físico e radiografia de tórax para avaliar o timo são fundamentais em pacientes com imunodeficiência combinada grave16.
Diagnóstico laboratorial
Cerca de 90% dos pacientes SCID são caracterizados por deficiência grave de linfócitos T, podendo apresentar ou não alterações no número absoluto de linfócitos B e células NK.
Linfopenia absoluta com valores < 2.500 linfócitos/mm3 de sangue (nível de referência normal no 1º ano de vida > 4.000 linfócitos/mm3 de sangue)17,18 é frequentemente vista em pacientes com essa imunodeficiência. Pacientes com SCID geralmente apresentam diminuição de linfócitos T CD3+ e podem apresentar hipogamaglobulinemia grave (IgG < 150 mg/dL)19, nem sempre observada pela presença de anticorpos maternos. Portanto, a realização de hemograma completo, contagem diferencial de leucócitos e medida de imunoglobulinas são importantes como investigação inicial da SCID, uma vez que linfopenia neonatal pode caracterizar a enfermidade2,20.
A realização de ensaios de imunofenotipagem por citometria de fluxo visando a identificação de subtipos de linfócitos em sangue periférico pode demonstrar a presença de fenótipos característicos para as várias formas da doença, quanto à presença de linfócitos T, B e células NK21. A caracterização de quais subpopulações celulares se encontram presentes ou ausentes (linfócitos T CD4+ e CD8+; linfócitos B CD19+; e células NK CD3-CD16/56+) possibilita o diagnóstico classificatório do tipo de SCID apresentada pelo paciente, auxiliando na identificação do possível defeito genético20.
A avaliação da função dos linfócitos T pode ser determinada in vitro pela avaliação de respostas celulares a mitógenos, tais como fito-hemaglutinina (PHA) e concanavalina A (ConA), a antígenos específicos, ou células alogênicas. Em SCID, a resposta de linfócitos T aos mitógenos está muito baixa ou ausente (< 5% de respostas em indivíduos saudáveis), sendo um elemento crucial para o diagnóstico de imunodeficiência combinada grave. As respostas à estimulação com antígenos específicos apresentam significância apenas após vacinação (tétano, tuberculina) ou a ocorrência de infecções (Candida, CMV ou Varicella zoster)15,2, sendo que este exame tem pouca ou nenhuma validade em crianças menores que 1 ano e especialmente nas menores de 6 meses. Tendo em vista que algumas formas de SCID apresentam defeitos na maturação dos linfócitos T (deficiência de ADA) ou a presença de mutações hipomórficas que promovem função residual de uma proteína defeituosa, a contagem absoluta de linfócitos pode apresentar valores próximos do limite inferior de normalidade e, na ausência de infecção, a contagem de neutrófilos é normal, podendo ocorrer eventual eosinofilia22.
Testes que avaliam o padrão de expressão e função da proteína γc ou JAK3 encontram-se disponíveis para ajudar no diagnóstico de imunodeficiência combinada grave. A confirmação do diagnóstico pode ser feita por ensaio de citometria de fluxo, por expressão anormal de γc ou JAK3 em células do sangue periférico. Ensaios específicos adicionais incluem immunoblotting para JAK3 e γc, detecção de IL-2 induzida por fosforilação de JAK3 e influxo de cálcio na ativação de células T CD4+; ensaios bioquímicos incluem a determinação do acúmulo de metabólitos tóxicos na deficiência de ADA23.
Nos primeiros meses de vida, a produção de anticorpos é reduzida ou ausente em pacientes com SCID, entretanto níveis séricos normais de IgG podem ser observados por transmissão materno-fetal. Há a possibilidade de identificação de redução significativa nos níveis de IgM. Avaliação detalhada da imunidade humoral por meio da quantificação de anticorpos específicos após vacinação, isohemaglutininas, ou subclasses de IgG, deve ser realizada e interpretada no contexto da idade do paciente, não sendo úteis antes do segundo ano de vida. Apesar disso, a realização destes exames em pacientes com suspeita de IDP envolvendo defeito na produção de anticorpos deve ser realizada2.
O diagnóstico precoce de SCID ainda é um desafio. A contagem diferencial de leucócitos em hemograma de recém-nascidos revelando linfopenia e a investigação de causas indeterminadas de linfopenia no período neonatal poderiam facilitar o diagnóstico precoce desta condição clínica, entretanto contagem normal de leucócitos no sangue periférico do recém-nascido pode ocorrer pela presença de células oriundas da circulação sanguínea materna2.
Diagnóstico genético-molecular
As estratégias utilizadas para a identificação de mutações causadoras de IDPs incluem, de forma resumida, três possibilidades: (1) estimativas baseadas em vias de sinalização conhecidas como essenciais para o desenvolvimento e função de células imunes, (2) semelhança de fenótipos clínicos com modelos murinos, e (3) abordagens genéticas imparciais.
Imunodeficiência combinada grave é uma doença com perfil genético heterogêneo, tendo sido identificadas numerosas mutações até o momento. O diagnóstico genético-molecular é direcionado por resultados de exames laboratoriais, e permite o diagnóstico definitivo. Além disso, é útil para estabelecer o diagnóstico nas apresentações atípicas, e fornecer informações relevantes quando há relação geno-fenotípica forte, propiciando avaliação de prognóstico e conduzindo a manejo adequado dos pacientes22,24.
O diagnóstico pré-natal de SCID pode ser feito in utero, particularmente quando existe criança afetada na família com defeito molecular identificado, podendo ser realizado a partir de células obtidas de vilosidades coriônicas ou por amniocentese. Após o nascimento, o diagnóstico pode ser confirmado através da obtenção de linfócitos T a partir do cordão umbilical25.
A avaliação do padrão de mutações cromossômicas por hibridização fluorescente in situ (FISH) é uma metodologia utilizada em alguns casos de investigação genético-molecular de pacientes com SCID, que possibilita avaliação do sucesso de transplante hematopoiético nesses pacientes. Estudos adicionais relatam o uso de FISH em paciente diagnosticado com pancitopenia, imunodeficiência combinada celular moderada com múltiplas anormalidades e retardo mental grave, indicando possível contribuição de alteração no gene NFRKB e no proto-oncogene ETS-1 nesta imunodeficiência26.
Quando comparado ao FISH e imunofenotipagem, o sequenciamento por método de Sanger se destaca como a metodologia mais apropriada para o diagnóstico molecular de SCID, uma vez que o sequenciamento de genes específicos possibilita a identificação da mutação genética responsável pela manifestação fenotípica da doença.
Os defeitos moleculares da SCID podem ser agrupados em quatro categorias distintas: (1) defeitos nos receptores de citocinas, como γc e IL7Rα; (2) defeitos em vias de sinalização das células T, como JAK3, CD45 e CD3; (3) defeitos na recombinação V(D)J, como RAG1/2; e (4) acúmulo de metabólitos tóxicos, como observado na deficiência de ADA27.
Mutações no gene IL2RG que codifica a cadeia γc se manifestam sob a forma de SCID-X, caracterizada pelo fenótipo T-B+NK-. Este perfil de linfócitos também é visto em formas autossômicas recessivas de SCID causadas por mutação no gene JAK328, que resulta na atuação deficiente da tirosina quinase intracelular necessária para a transdução de sinal através de receptores de citocinas da γc. Por conta dessa característica, as deficiências γc e JAK3 são consideradas indistinguíveis do ponto de vista clínico e imunológico.
É de conhecimento que a presença de heterogeneidade dos defeitos genéticos ao longo de uma mesma via de sinalização faz um paralelo com a heterogeneidade de fenótipos imunológicos29. Sendo assim, o diagnóstico molecular de SCID para os casos de SCID-X e de defeitos de JAK3 é feito com base em várias estratégias: caracterização das propriedades funcionais da sinalização via γc/JAK3; imunofenotipagem de células mononucleares do sangue periférico; análise da proliferação de células NK e atividade citolítica; identificação de mutações nos genes da γc e JAK3 por sequenciamento do DNA genômico28. Além disso, quando há a presença de resultados sugestivos, a avaliação de splicing aberrante de mRNA pode ser realizado por RT-PCR e por subsequente clonagem e sequenciamento dos produtos da RT-PCR30. Outras possibilidades de análise incluem o rastreio de DNA genômico utilizando análise de polimorfismo conformacional de cadeia simples (SSCP), seguido de sequenciamento dos exons que apresentam padrão alterado28.
O diagnóstico das diversas formas de SCID como as associadas a defeitos em JAK3, IL7Rα, CD45, e CD3 (cadeias γ, δ e ε) juntamente com a cadeia ζ, bem como de casos de SCID-X, é baseado no fenótipo imune, sendo confirmado por diagnóstico molecular através do sequenciamento de genes31-33. Os casos de deficiência de ADA são diagnosticados por meio de avaliação dos níveis de ADA ou de seus metabólitos em células de pacientes com fenótipo SCID T-B-NK-, tendo sua confirmação feita também através da identificação da mutação34.
As células T maternas alorreativas podem levar a quadro clínico de "doença do enxerto-versus-hospedeiro" (GVHD) materno-fetal, geralmente condição fatal que ocorre em pacientes com imunodeficiência combinada grave. Para o diagnóstico, a suspeita da presença de células T maternas deve ser eliminada através de análises de quimerismo: em pacientes do sexo masculino por meio de hibridização in situ XX/XY em células CD3+, e em pacientes do sexo feminino pela técnica da PCR do DNA, através de análises do HLA ou de repetição em tandem em número variável (VNTR) utilizando células CD3+ obtidas a partir de células mononucleares do sangue periférico (PBMC), ou a partir de biópsia da pele, para análise do cromossomo Y. Quando presente, indica ausência da GVHD, confirmando presença de Síndrome de Omenn nesses pacientes. Quando ausente, indica doença do enxerto-versus-hospedeiro. O cromossomo Y é útil na investigação para a diferenciação de GVHD materno-fetal e Síndrome de Ommen em pacientes que apresentam diagnóstico indefinido. Em todos os casos, infecções por HIV devem ser excluídas35. Em casos de necessidade de transfusão sanguínea ou plaquetas, o recém-nascido deve receber sangue total ou plaquetas irradiadas (CMV negativo, sem leucócitos), para prevenir GVHD e risco de contaminação por CMV.
Com o advento de novas tecnologias e devido ao grande número de genes envolvidos na SCID, as mutações passaram também a ser investigadas utilizando-se tecnologias de arranjo (biochips de DNA ou cDNA), que possibilitam a confirmação de aproximadamente 90% das mutações ligadas ao cromossomo X, além de apresentar melhor relação custo-benefício em relação ao sequenciamento Sanger. No entanto, estimativas precisas para a SCID por deficiência de IL7R autossômica e JAK3 são difíceis devido ao baixo número de mutações descritas. Além das opções de arranjos para a identificação de mutações, o uso de novas tecnologias como o sequenciamento de nova geração (NGS) tem sido avaliado para a identificação de mutações de forma eficiente em amostras de DNA de genomas inteiros ou de exomas36. Avanços recentes possibilitam solucionar casos dificeis em doenças com formas de herança autossômica dominante, penetrância incompleta ou mutações em regiões não codificantes37,38.
Triagem neonatal
A triagem neonatal tem o objetivo de realizar diagnóstico precoce de diversas doenças congênitas ou infecciosas no período neonatal, permitindo que o tratamento adequado seja instituído39. Na década de 1960, o Dr. Robert Guthrie desenvolveu um ensaio para triagem neonatal em larga escala da fenilcetonúria40, utilizando um sistema de coleta e transporte de amostras de sangue em papel de filtro41. Em 2001 com a criação do Programa Nacional de Triagem Neonatal (PNTN), além do diagnóstico da fenilcetonúria, outras doenças como hipotireoidismo congênito, fibrose cística e hemoglobinopatias passaram a ser incluídas no programa de triagem42.
Em 2008, o estado americano de Wisconsin foi pioneiro em instituir triagem neonatal para SCID43, seguido pelos estados de Massachusetts e California. A triagem de SCID é feita através da quantificação do número de círculos de excisão em receptor de linfócitos T (do inglês: T Cell Receptor Excision Circles - TRECs), que são marcadores do desenvolvimento normal deste tipo celular. Amostras de sangue são colhidas de recém-nascidos em cartões de Guthrie (papel de filtro)44, e aquelas com baixo número ou níveis indetectáveis de TRECs são características de SCID e de outras condições em que a produção e/ou sobrevivência das células T está profundamente prejudicada. Como os linfócitos T estão reduzidos em todas as formas de SCID e os TRECs não se replicam com a divisão celular, o ensaio de quantificação de TRECs serve como biomarcador para o número de células T que emergiram do timo, possibilitando a identificação de crianças com SCID independentemente do defeito molecular, ao apresentarem baixos níveis de TRECs. Para melhorar a precisão da triagem neonatal, a dosagem concomitante de interleucina-7 (IL-7) a partir de cultura de PBMC é recomendada, uma vez que esta citocina está associada ao desenvolvimento de células T. Combinando estes dois testes, a sensibilidade do diagnóstico é próxima a 100%45-47.
Além da análise de TRECs para os linfócitos T, existe também o KREC (do inglês kappa deleting recombination excision circles - círculos de excisão de recombinação de deleção de kappa) que também é um segmento de DNA circular. No entanto, este é gerado durante a maturação de linfócitos B na medula óssea. Durante a maturação de linfócitos B, os KRECs são formados e os produtos dos eventos de recombinação que determinam a exclusão alélica e isotípica do locus da imunoglobulina kappa (IgK), o tornam não funcional48. Este produto, que funciona como biomarcador da imunidade de linfócitos B, quando quantificado, permite uma melhor caracterização das formas de SCID, um acompanhamento do transplante alogênico de células-tronco hematopoiéticas (TCTH) e das terapias de reposição enzimática. Valores < 10 KRECs/µL de sangue são considerados baixos49.
Atualmente, SCID passou a ser reconhecida como uma doença que atende os critérios de inclusão em Programa Nacional de Triagem Neonatal. As estimativas apontavam que 50% das crianças acometidas por SCID evoluíam para óbito por atraso no diagnóstico. Resultados da triagem neonatal apontam para impacto positivo e significante na sobrevivência de portadores de SCID, revelando que 85% das crianças testadas ao nascimento sobreviveram, quando comparadas a 58% das crianças não testadas50,51. O ensaio de TREC tem demonstrado ser um teste com excelente especificidade e sensibilidade para identificar crianças com SCID e outras formas de linfopenia de linfócitos T de forma precisa, e já foi integrado de forma eficiente a programas de saúde publica em alguns estados dos Estados Unidos e Canadá, como por exemplo Wisconsin, California, New York, e Ontario. O custo de saúde de apenas uma criança com SCID pode ser maior que o custo da triagem da doença para uma região populacional inteira52. Baseada nestes dados, a Jeffrey Modell Foundation (JMF), fundação que possui uma longa história de incentivo para a implementação da triagem neonatal no mundo, desenvolveu um algoritmo ou "árvore de decisão", validado por dados da literatura científica, a ser usado por departamentos da saúde pública e ministérios de saúde ao redor do mundo. Essa ferramenta de "decisão" permite que dados locais ou regionais possam ser usados para medir o impacto econômico da implementação da triagem neonatal para SCID e linfopenias de linfócitos T53.
No Brasil, a triagem neonatal, conhecida popularmente como Teste do Pezinho, teve início em 1976, com o projeto do Prof. Benjamin Schmidt para a triagem de fenilcetonúria54 e atualmente conta com iniciativas de projetos em desenvolvimento que visam contribuir para o avanço na implementação de testes de triagem neonatal para as IDPs, especificamente SCID, além de ajudar na estimativa da sua real incidência na população brasileira55.
TRATAMENTO
SCID foi a primeira doença cujo tratamento foi realizado através do TCTH com sucesso. Através desse procedimento, pode-se restaurar o desenvolvimento de células T56. Atualmente, os imunologistas clínicos incluem o TCTH como tratamento primordial (padrão ouro), curativo, para pacientes com imunodeficência combinada grave. Pacientes apresentam uma alta taxa de sobrevida (70-96%) e cura quando o TCTH é realizado logo após o nascimento, em especial até os cinco primeiros meses de vida. Pacientes com SCID não tratados raramente sobrevivem além dos 6-12 meses de vida57. Até a realização do transplante, algumas medidas são necessárias: (1) isolamento da criança, evitando contato com outras crianças e potenciais contatos contagiosos; (2) reposição de imunoglobulina humana; (3) profilaxia para Pneumocystis jirovecii, vírus respiratório sincicial; (4) não administração de vacinas de patógenos atenuados; (5) irradiação de todos os hemoderivados.
Após transplante bem sucedido, a maioria dos pacientes apresenta pleno funcionamento do sistema imune, sendo ocasionalmente necessária terapia de reposição de imunoglobulina humana. Apesar de o TCTH ser a melhor opção de tratamento, ressalta-se que a mortalidade relacionada ao procedimento é elevada quando há a presença de sequelas de infecções em órgãos como cérebro, pulmão ou fígado58.
Para crianças com SCID ocasionada por deficiência de ADA, existe um estudo clínico de terapia de substituição enzimática, com aplicação semanal de ADA conjugada ao polietineloglicol (PEG-ADA) por injeção intramuscular. Com esta terapia mantém-se a atividade da ADA de forma elevada no plasma e, como consequência, a função imune é reconstituída após 2-4 meses de terapia34.
Outra forma de tratamento inclui a terapia gênica, que tem apresentado resultados promissores para crianças com SCID por deficiência no gene ADA, IL-2R e nos casos de SCID-X. Com a terapia gênica, há a introdução de uma cópia normal do gene mutado em substituição ao gene defeituoso por intermédio de vetores retrovirais, corrigindo assim o fenótipo da doença. Da mesma forma que para o TCTH, a possibilidade de início do tratamento por meio de terapia gênica deve ser analisada por especialistas da área e iniciada o mais cedo possível37,59-61.
CONCLUSÕES
Imunodeficiência combinada grave é uma IDP que compreende um grupo heterogêneo de defeitos genético-moleculares, caracterizada por comprometimento marcante da imunidade, podendo levar o paciente a óbito cedo na infância por infecções graves se a enfermidade não for diagnosticada e tratada precocemente21.
O diagnóstico dos diversos fenótipos clínicos e imunológicos da SCID é feito com base na história clínica e exame físico, e em exames laboratoriais. Métodos genético-moleculares podem possibilitar a identificação da mutação causadora da doença10.
Melhora no conhecimento das apresentações clínicas da doença e recentes avanços em diagnóstico genético-molecular e em modalidades terapêuticas têm permitido a identificação precoce e tratamento eficaz da imunodeficiência combinada grave. O tratamento com TCTH é considerado essencial para o reestabelecimento do sistema imune de pacientes com imunodeficiência combinada grave. Melhores taxas de sobrevida são obtidas se o TCTH for realizado antes dos primeiros 3-5 meses de vida62. A perspectiva de utilização de novas medidas terapêuticas como a terapia gênica somática tem sido promissora.
REFERÊNCIAS
1. Buckley RH. The long quest for neonatal screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129:597-604.
2. Buckley RH. Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004;22:625-55.
3. Roifman CM, Somech R, Kavadas F, Pires L, Nahum A, Dalal I, et al. Defining combined immunodeficiency. J Allergy Clin Immunol. 2012;130:177-83.
4. Glanzmann E, Riniker P. Essential lymphocytophthisis; new clinical aspect of infant pathology. Ann Paediatr. 1950;175:1-32.
5. Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the international union of Immunological societies expert committee for primary immunodeficiency. Front Immunol. 2014;5:162.
6. Lindegren ML, Kobrynski L, Rasmussen SA, Moore CA, Grosse SD, Vanderford ML, et al. Applying public health strategies to primary immunodeficiency diseases: a potential approach to genetic disorders. MMWR Recomm. 2004;53:1-29.
7. Dornas PB, Robazzi TC, Silva LR. Imunodeficiência primária: quando investigar, como diagnosticar. Pediatria (São Paulo). 2010;32:51-62.
8. Errante PR, Franco JL, Espinosa-Rosales FJ, Sorensen R, Condino-Neto A. Advances in primary immunodeficiency diseases in Latin America: epidemiology, research, and perspectives. Ann Acad Sci. 2012;1250:62-72.
9. Mazzucchelli JT, Bonfim C, Castro GG, Condino-Neto AA, Costa NM, Cunha L, et al. Severe combined immunodeficiency in Brazil: management, prognosis, and BCG-associated complications. J Investig Allergol Clin Immunol. 2014;24:184-91.
10. Cowan MJ, Logan BR, Notarangelo LD, Griffith LM, Puck JM, et al. The natural history of children with severe combined immunodeficiency: baseline features of the first fifty patients of the primary immune deficiency treatment consortium prospective study 6901. J Clin Immunol. 2013;33:1156-64.
11. Stocks RM, Thompson JW, Church JA, Kun S, Simms E. Severe combined immunodeficiency: otolaryngological presentation and management. Ann Otol Rhinol Laryngol. 1999;108:403-7.
12. Walzer PD, Schultz MG, Western KA, Robbins JB. Pneumocystis carinii pneumonia and primary immune deficiency diseases of infancy and childhood. J Pediatr. 1973;82:416-22.
13. Abramowsky C, Gonzales B, Sorensen RU. Disseminated bacillus Calmette-Guérin infections in patients with primary immunodeficiencies. Am J Clin Pathol. 1993;100:52-6.
14. Antaya RJ, Gardner ES, Bettencourt MS, Daines M, Denise Y, Uthaisangsook S, et al. Cutaneous complications of BCG vaccination in infants with immune disorders: two cases and a review of the literature. Pediatric Dermatol. 2001;18:205-9.
15. Patel DD, Gooding ME, Parrott RE, Curtis KM, Haynes BF, Buckley RH. Thymic function after hematopoietic stem-cell transplantation for the treatment of severe combined immunodeficiency. N Engl J Med. 2000;342:1325-32.
16. Myers LA, Patel DD, Puck JM, Buckley RH. Hematopoietic stem cell transplantation for severe combined immunodeficiency in the neonatal period leads to superior thymic output and improved survival. Blood. 2002;99:872-8.
17. Hernandez-LLizaliturri FJ. B-cell and T-cell combined disorders. Emedicine 2008 Jun 24. Disponível em: http://www.emedicine.com/med/topic3538.htm.
18. El-Sayed SS, Afifi HMES, Yahia AS. Neonatal screening for absolute lymphopenia. Egypt J Pediatr Allergy Immunol. 2013;11:75-81.
19. Elder ME. T-cell immunodeficiencies. Pediatr Clin North Am. 2000;47:1253-74.
20. Rosen FS. Defects in cell-mediated immunity. Clin Immunol Immunopathol. 1986;41:1-7.
21. Gaspar HB, Gilmour KC, Jones AM. Severe combined immunodeficiency-molecular pathogenesis and diagnosis. Arch Dis Child. 2001;84:169-73.
22. Platt C, Geha RS, Chou J. Gene hunting in the genomic era: Approaches to diagnostic dilemmas in patients with primary immunodeficiencies. J Allergy Clin Immunol. 2014;134:262-8.
23. Puck JM, Pepper AE, Henthorn PS, Candotti F, Isakov J, Whitwam T, et al. Mutation analysis of IL2RG in human X-linked severe combined immunodeficiency. Blood. 1997;89:1968-77.
24. Notarangelo LD, Sorensen R. Is it necessary to identify molecular defects in primary immunodeficiency disease? J Allergy Clin Immunol. 2008;122:1069-73.
25. Buckley RH. Primary immunodeficiency diseases: dissectors of the immune system. Immunol Rev. 2002;185:206-19.
26. Bielorai B, Trakhtenbrot L, Amariglio N, Rothman R, Tabori U, Dallal I, et al. Multilineage hematopoietic engraftment after allogeneic peripheral blood stem cell transplantation without conditioning in SCID patients. Bone Marrow Transplant. 2004;34:317-20.
27. Liston A, Enders A, Siggs OM. Unravelling the association of partial T-cell immunodeficiency and immune dysregulation. Nat Rev Immunol. 2008;8:545-58.
28. Gilmour KC, Cranston T, Loughlin S, Gwyther J, Lester T, Espanol T, et al. Rapid protein-based assays for the diagnosis of T-B+ severe combined immunodeficiency. Br J Haematol. 2001;112:671-6.
29. Notarangelo LD, Giliani S, Mella P, Schumacher RF, Mazza C, Savoldi G, et al. Combined immunodeficiencies due to defects in signal transduction: defects of the gammac-JAK3 signaling pathway as a model. Immunobiology. 2000;202:106-19.
30. Ginn SL, Smyth C, Wong M, Bennetts B, Rowe PB, Alexander IE. A novel splice-site mutation in the common gamma chain (gammac) gene IL2RG results in X-linked severe combined immunodeficiency with an atypical NK+ phenotype. Hum Mutat. 2004;23:522-3.
31. Roifman CM, Zhang J, Chitayat D, Sharfe N. A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood. 2000;96:2803-7.
32. Roberts JL, Buckley RH, Luo B, Pei J, Lapidus A, Peri S, et al. CD45-deficient severe combined immunodeficiency caused by uniparentaldisomy. Proc Natl Acad Sci USA. 2012;109:10456-61.
33. Recio MJ, Moreno-Pelayo MA, Kiliç SS, Guardo AC, Sanal O, Allende LM, et al. Differential biological role of CD3 chains revealed by human immunodeficiencies. J Immunol. 2007;178:2556-64.
34. Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114:3524-32.
35. Appleton AL, Curtis A, Wilkes J, Cant AJ. Differentiation of materno-fetal GVHD from Omenn's syndrome in pre-BMT patients with severe combined immunodeficiency. Bone Marrow Transplant. 1994;14:157-9.
36. Ghosh S, Krux F, Binder V, Gombert M, Niehues T, Feyen O, et al. Array-Based Sequence Capture and Next-Generation Sequencing for the Identification of Primary Immunodeficiencies. Scand J Immunol. 2012;75:350-4.
37. van der Burg M, van Zelm MC, van Dongen JJ. Molecular diagnostics of primary immunodeficiencies: benefits and future challenges. Adv Exp Med Biol. 2009;634:231-41.
38. McCabe LL, McCabe ER. Expanded newborn screening: implications for genomic medicine. Annu Rev Med. 2008;59:163-75.
39. Routes JM, Grossman WJ, Verbsky J, Laessig RH, Hoffman GL, Brokopp CD, et al. Statewide newborn screening for severe T-cell lymphopenia. JAMA. 2009;302:2465-70.
40. Guthrie R, Susi I. A simple phenylalanine method for detecting phenylketonuria in large populations of newborn infants. Pediatrics. 1963;32:318-43.
41. Lesser AJ. Phenylketonuria and the Guthrie test. Pediatrics. 1963;32:940.
42. Baker MW, Grossman WJ, Laessig RH, Hoffman GL, Brokopp CD, Kurtycz DF, et al. Development of a routine newborn screening protocol for severe combined immunodeficiency. J Allergy Clin Immunol. 2009;124:522-7.
43. Verbsky J, Thakar M, Routes J. The Wisconsin approach to newborn screening for severe combined immunodeficiency. J Allergy Clin Immunol. 2012;129:622-7.
44. Chase NM, Verbsky JW, Routes JM. Newborn screening for T-cell deficiency. Curr Opin Allergy Clin Immunol. 2010;10:521-5.
45. van Zelm MC, van der Burg M, Langerak AW, van Dongen JJ. PID comes full circle: applications of V(D)J recombination excision circles in research, diagnostics and newborn screening of primary immunodeficiency disorders. Front Immunol. 2011;2:12.
46. Morinish Y, Imai K, Nakagawa N, Sato H, Horiuchi K, Ohtsuka Y, et al. Identification of severe combined immunodeficiency by T-cell receptor excision circles quantification using neonatal Guthrie cards. J Pediatr. 2009;155:829-33.
47. McGhee SA, Stiehm ER, Cowan M, Krogstad P, McCabe ER. Two-tiered universal newborn screening strategy for severe combined immunodeficiency. Mol Genet Metabol. 2005;86:427-30.
48. van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJ. Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J Exp Med. 2007;204:645-55.
49. Nakagawa N, Imai K, Kanegane H, Sato H, Yamada M, Kondoh K, et al. Quantification of κ-deleting recombination excision circles in Guthrie cards for the identification of early B-cell maturation defects. J Allergy Clin Immunol. 2011;128:223-5.
50. Brown L, Xu-Bayford J, Allwood Z, Slatter M, Cant A, Davies EG, et al. Neonatal diagnosis of severe combined immunodeficiency leads to significantly improved survival outcome: the case for newborn screening. Blood. 2011;117:3243-6.
51. PuckJM. Neonatal screening for severe combined immune deficiency. Curr Opin Allergy Clin Immunol. 2007;7:522-7.
52. Modell V, Knaus M, Modell F. An analysis and decision tool to measure cost benefit of newborn screening for severe combined immunodeficiency (SCID) and related T-cell lymphopenia. Immunol Res. 2014;60:145-52.
53. Kwan A, Church JA, Cowan MJ, Agarwal R, Kapoor N, Kohn DB, et al. Newborn screening for severe combined immunodeficiency and T-cell lymphopenia in California: results of the first 2 years. J Allergy Clin Immunol. 2013;132:140-50.
54. São Paulo. Lei Estadual nº 3.914, de 14 de novembro de 1983. Disponível em: http://www.jusbrasil.com.br/legislacao/197755/ lei-3914-83-sao-paulo-sp. Acessado em 13/fev/2011.
55. Kanegae MPP, Santos AMN, Cavalcanti CM, Neto AC. Triagem neonatal para imunodeficiência combinada grave. Rev bras alergia imunopatol. 2011;34:7-11.
56. Antoine C, Müller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J et al. Long-term survival and transplantation of hematopoietic stem cells for immunodeficiencies: report of the European experience 1968-99. Lancet. 2003;361:553-60.
57. Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, e tal. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014;371:434-46.
58. Brasil. Ministério da Saúde. Portaria nº. 822, de 06 de junho de 2001 [Internet]. Disponível em: http://dtr2001.saude.gov.br/sas/PORTARIAS/Port2001/GM/GM-822.htm. Acessado: 05 de setembro de 2012.
59. Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy for primary adaptive immune deficiencies. J Allergy Clin Immunol. 2011;127:1356-9.
60. Qasim W, Gennery AR. Gene Therapy for Primary Immunodeficiencies: Current Status and Future Prospects. Drugs. 2014;74:963-9.
61. Touzot F, Hacein-Bey-Abina S, Fischer A, Cavazzana M. Gene therapy for inherited immunodeficiency. Expert Opin Biol Ther. 2014;14:789-98.
62. Rezaei N, Notarangelo L, Aghamohammadi A, editors. Primary Immunodeficiency Diseases: Definition, Diagnosis, and Management. Ed. Springer, 2008.
63. Al-Herz W, Bousfiha A, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54.
64. Notarangelo LD. Primary immunodeficiencies. J Allergy Clin Immunol. 2010;125:182-94.
65. Leung DYM, Sampson Ha, Geha R, Szefler SJ. Severe combined immunodeficiency. Pediatric Allergy: Principles and practice [Internet]. Ed. Elsevier; 2a ed. Capítulo 9. Disponível em: https://www.inkling.com/read/pediatric-allergy-principles-practice-leung-2nd/chapter-9/severe-combined-immunodeficiency.
66. Pannicke U, Hönig M, Hess I, Friesen C, Holzmann K, Rump EM, et al. Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Gen. 2008;41:101-5.
67. Rezaei N, Aghamohammadi A, Notarangelo LD. Primary immunodeficiency diseases. Definition, diagnosis and management [Internet]. Ed. Springer; 2008. Capítulo 2. Disponível em: http://www.beck-shop.de/fachbuch/leseprobe/9783540785378_Excerpt_001.pdf
68. Borte S, Wang N, Oskarsdóttir S, von Döbeln U, Hammarström L. Newborn screening for primary immunodeficiencies: beyond SCID and XLA. Ann NY Acad Sci. 2011;1246:118-30.
69. Butte MJ, Haines C, Bonilla FA, Puck J. IL-7 receptor deficient SCID with a unique intronic mutation and post-transplant autoimmunity due to chronic GVHD. Clin Immunol. 2007;12:159-64.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888