Número Atual: Janeiro-Fevereiro 2014 - Volume 2 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
Angioedema hereditário e outras formas de angioedema por bradicinina: atualização no diagnóstico e tratamento
Hereditary angioedema and other forms of bradykinin-mediated angioedema: update on diagnosis and treatment
Maria Fernanda Ferraro1; L. Karla Arruda2; Luana S. M. Maia3; Adriana S. Moreno4
1. MD, PhD. Universidade Presidente Antônio Carlos (Unipac), Araguari, MG
2. MD, PhD. Faculdade de Medicina de Ribeirao Preto, Universidade de Sao Paulo
3. MSc. Faculdade de Medicina de Ribeirao Preto, Universidade de Sao Paulo
4. PhD. Faculdade de Medicina de Ribeirao Preto, Universidade de Sao Paulo
Endereço para correspondência:
Luisa Karla de Paula Arruda
E-mail: karla@fmrp.usp.br
Submetido em 12/05/2015.
Aceito em 16/05/2015.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
Angioedema é definido como edema que ocorre em áreas bem delimitadas do tecido subcutâneo e submucoso, consequente ao aumento da permeabilidade capilar local causada por mediadores vasoativos. Em geral acomete extremidades, face, vias aéreas superiores e tratos gastrointestinal e geniturinário. Há dois mediadores amplamente reconhecidos na patogênese do angioedema: histamina e bradicinina, com repercussoes clínicas distintas. O angioedema histaminérgico é geralmente associado a urticária e tem boa resposta ao tratamento com anti-histamínicos, corticosteroides e adrenalina. Já o angioedema por bradicinina tem duraçao mais prolongada (36 a 72 horas), envolve mais frequentemente o trato gastrointestinal e nao responde ao tratamento convencional com anti-histamínicos, corticosteroides e adrenalina. Suspeita-se de angioedema mediado por bradicinina quando há angioedema recorrente, nao associado a urticária, que pode ser hereditário ou adquirido. O nonapeptídeo bradicinina é um potente mediador de vasodilataçao e aumento da permeabilidade vascular, particularmente em vênulas pós-capilares, que produz seus efeitos através da estimulaçao dos receptores B2 ligados a proteína-G. Neste artigo de revisao, temos por objetivo fazer uma atualizaçao em diagnóstico diferencial e tratamento das diferentes formas de angioedema mediado por bradicinina: angioedema por inibidores da enzima conversora da angiotensina (iECA) e outros fármacos que podem diminuir o metabolismo da bradicinina; angioedema hereditário (AEH) por mutaçoes nos genes SERPING1 e F12 que codificam, respectivamente, o inibidor de C1-INH e o fator XII da coagulaçao; angioedema hereditário de causa desconhecida; angioedema por deficiência adquirida do C1-INH; e angioedema idiopático.
Descritores: Angiodema, angioedema hereditário, bradicinina, inibidor de C1, angioedema adquirido.
INTRODUÇÃO
Angioedema é definido como edema que ocorre em áreas bem delimitadas do tecido subcutâneo e submucoso, consequente ao aumento da permeabilidade capilar local causada por mediadores vasoativos. O angioedema frequentemente acomete as extremidades, a face, as vias aéreas superiores e os tratos gastrointestinal e geniturinário. Mais comumente ocorre como manifestação de doenças alérgicas e de diferentes formas de urticária, mas existem situações nas quais o angioedema representa uma doença em si.
O angioedema pode se constituir em uma emergência médica. Quando acomete a cavidade oral, particularmente língua, orofaringe e laringe, pode causar asfixia e morte. O envolvimento do trato gastrointestinal por angioedema pode levar a episódios de dor abdominal intensa, grave, acompanhada de ascite, náuseas, vômitos e diarreia, que podem requerer tratamento hospitalar. Além disso, condições que levam a episódios recorrentes de angioedema geram um comprometimento marcante na qualidade de vida dos indivíduos acometidos.
Há dois mediadores amplamente reconhecidos na patogênese do angioedema: a histamina e a bradicinina, com algumas repercussões clínicas distintas1. O angioedema histaminérgico pode apresentar hiperemia e prurido local, mas a característica mais marcante (que o distingue da forma mediada por bradicinina) é a associação com urticária na maioria dos casos, e a boa resposta ao tratamento com anti-histamínicos, corticosteroides e adrenalina. Por outro lado, pode-se considerar como características peculiares do angioedema por bradicinina a duração mais prolongada dos episódios (36 a 72 horas, enquanto a forma histaminérgica costuma ter resolução espontânea em até 24 horas) e também o envolvimento muito mais frequente do trato gastrointestinal2. É importante também o fato de que, pela própria patogênese, este tipo de angioedema não apresenta boa resposta ao tratamento convencional com anti-histamínicos, corticosteroides e adrenalina.
Angioedema recorrente não associado a urticária induz à suspeita de angioedema mediado por bradicinina, que pode ser hereditário ou adquirido. O nonapeptídeo bradicinina é um potente mediador de vasodilatação e aumento da permeabilidade vascular, particularmente em vênulas pós-capilares, que produz seus efeitos através da estimulação dos receptores B2 ligados à proteína-G (B2R). A bradicinina foi descoberta pelo médico e cientista brasileiro Dr. Mauricio Rocha e Silva, no curso de experimentos conduzidos no Instituto Biológico em São Paulo, ao investigar o papel fisiológico do veneno de cobra jararaca Bothrops jararaca3. Estudos subsequentes desenvolvidos por Rocha e Silva e Sérgio Ferreira na Faculdade de Medicina de Ribeirão Preto resultaram na identificação de uma fração peptídica do veneno de Bothrops jararaca que potencializava a ação da bradicinina em vários órgãos e na pressão arterial de gatos. Os autores demonstraram que esse fator, denominado Bradykinin Potentiating Factor, causava inibição da ação de enzimas que degradam bradicinina4. Essas descobertas por pesquisadores brasileiros forneceram a base para o desenvolvimento dos inibidores da enzima conversora da angiotensina (iECA), introduzidos em 1981, que são medicações amplamente utilizadas no tratamento da hipertensão arterial e insuficiência cardíaca congestiva em todo o mundo, com grande eficácia. De forma interessante, um dos efeitos adversos do uso de iECA é o aparecimento de angioedema que ocorre em cerca de 0,7% dos pacientes, e que se assemelha ao angioedema hereditário em muitos de seus aspectos clínicos. O mecanismo principal do angioedema por iECA, que é classe-específico, é atribuído a aumento da bradicinina por diminuição da degradação da mesma pela ECA e possivelmente deficiência quantitativa e/ou qualitativa de outras proteases alternativas que têm ação na degradação da bradicinina, incluindo aminopeptidase P (APP) e dipeptidil peptidase IV (DPPIV)5.
O foco deste artigo é revisar o diagnóstico diferencial e o tratamento das diferentes formas de angioedema mediado por bradicinina: angioedema por iECA e outros fármacos que podem diminuir o metabolismo da bradicinina; angioedema hereditário (AEH) por mutações nos genes SERPING1 e F12 que codificam, respectivamente, o inibidor de C1 (C1-INH) e o fator XII da coagulação; angioedema hereditário de causa desconhecida; angioedema por deficiência adquirida do C1-INH; e angioedema idiopático.
ANGIOEDEMA INDUZIDO POR INIBIDORES DA ENZIMA CONVERSORA DA ANGIOTENSINA (iECA) E OUTROS FÁRMACOS COM EFEITOS NO CATABOLISMO DA BRADICININA
O primeiro passo na avaliação de pacientes com angioedema recorrente não associado a urticária é uma história detalhada sobre o uso de fármacos.
Os inibidores da enzima conversora da angiotensina (iECA) são drogas amplamente prescritas para o controle da pressão arterial e proteção renal. A enzima conversora da angiotensina é uma dipeptil carboxipeptidase que cliva certos peptídeos, incluindo a bradicinina e substância P. Com a sua inibição, a degradação dessas substâncias se torna mais lenta, contribuindo para ocorrência de angioedema mediado por bradicinina. Medicamentos da classe dos iECA devem ser suspensos em todos os pacientes com angioedema recorrente, mesmo que os episódios de angioedema tenham início anos depois da introdução da medicação.
Até 0,7% dos indivíduos que utilizam iECA apresentam angioedema recorrente1,6,7. Afro-americanos têm risco até 5 vezes maior que brancos para angioedema induzido por iECA. Outros fatores de risco são tabagismo, idade avançada e sexo feminino. Pacientes diabéticos têm menor risco que não diabéticos8.
O angioedema induzido por iECA tem predileção pela face, pescoço, língua, boca e laringe, mas existem casos esporádicos de envolvimento gastrointestinal e de extremidades. O tempo médio para início dos sintomas é de 1,8 anos após início do uso de iECA, embora em 25% dos casos os sintomas apareçam já no primeiro mês e em alguns pacientes apenas após 10 anos de uso. O diagnóstico é clínico, não existem dados laboratoriais que confirmem o diagnóstico. A remissão das crises de angioedema após a suspensão da medicação confirma o diagnóstico, mas isso pode levar, em média, 6 semanas. Embora o angioedema induzido por iECA possa apresentar sintomas similares aos do AEH, esses pacientes terão níveis antigênicos e funcionais do inibidor de C1 e de C4 normais.
Os bloqueadores dos receptores da angiotensina II (BRAs), anti-hipertensivos classicamente utilizados como alternativa aos iECA, podem também levar à ocorrência de angioedema, embora numa frequência bem menor9. Estima-se que menos de 10% dos pacientes com angioedema por iECA apresentarão angioedema também com BRAs (Figura 1). Angioedema associado aos BRAs é descrito como menos grave, e ocorre mais precocemente quando comparado ao angioedema que se desenvolve durante a terapia com iECA10.
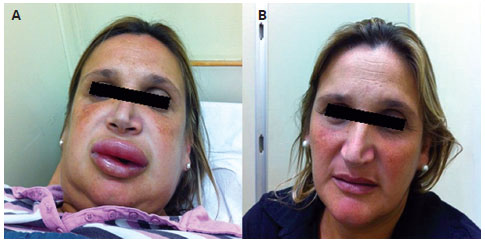
Figura 1 - Angioedema por bloqueador de receptores da angiotensina II (BRA). A: paciente com angioedema por valsartan; havia apresentado previamente angioedema recorrente, associado ao uso de enalapril. B: paciente após uma semana do episódio agudo
Outro anti-hipertensivo, inibidor da renina, o alisquireno também é associado à ocorrência de angioedema em cerca de 0,4% dos pacientes que usam esta medicação. Estudo de Toh et al. mostrou risco menor de angioedema com uso de BRA que com iECA ou alisquireno. Um total de 4.511 episódios de angioedema (3.301 por iECA, 288 por BRA, 7 por alisquireno) foram avaliados com hazard ratios ajustadas de 3,04 (95% intervalo de confiança [IC] 2,81-3,27) para iECA, 1,16 (95% IC 1,00-1,34) para BRA e 2,85 (95% IC 1,34-6,04) para alisquireno9.
O uso de omapatrilat, uma droga que inibe três enzimas responsáveis pela degradação da bradicinina (ECA, neprilisina e aminopeptidase P), para tratar pacientes com insuficiência cardíaca, esteve associado a angioedema grave em estudos clínicos. Neprilisina é uma endopeptidase neutra que degrada vários peptídeos vasoativos endógenos, incluindo a bradicinina. Estudo recente mostrou que a associação de inibidor de neprilisina e BRA (valsartan) em um fármaco denominado LCZ696 foi mais eficaz para o tratamento da insuficiência cardíaca, e não esteve associado ao maior risco de angioedema grave quando comparado ao tratamento com enalapril11.
Recentemente hipoglicemiantes da família das gliptinas (inibidores da dipeptil peptidase VI) têm sido associados à ocorrência de angioedema. Essa enzima tem um papel secundário no metabolismo da substância P e da bradicinina, assim a droga inibidora leva ao aumento dos níveis dessas substâncias. Risco aumentado de angioedema se dá especialmente naqueles pacientes já em uso de iECA, fato comum em pacientes com diabetes melittus12,13.
As enzimas responsáveis pelo catabolismo da bradicinina e seu metabólito desarginina-bradicinina, e os sítios de inibição de fármacos associados à ocorrência de angioedema estão indicados de forma esquemática na Figura 2.
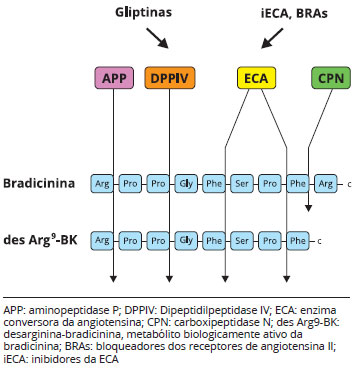
Figura 2 - Catabolismo das cininas e angioedema. Local de atuação de fármacos que inibem enzimas responsáveis pelo catabolismo da bradicinina
Pacientes com angioedema recorrente não associado a urticária e que não usem iECA ou outras drogas suspeitas devem ser avaliados através de história familiar detalhada e investigados para angioedema hereditário mediado por bradicinina (AEH tipos I e II, e AEH com C1-INH normal), e para angioedema por deficiência adquirida do inibidor de C1 (AEA).
ANGIOEDEMA HEREDITÁRIO
Angioedema hereditário (AEH) é uma doença de herança autossômica dominante, e em sua forma clássica ocorre devido a mutações no gene que codifica o inibidor de C1, designado como SERPING1. A prevalência média é de 1 caso para cada 50.000 pessoas. As anormalidades estruturais do gene nesses pacientes são muito heterogêneas, e já foram descritas cerca de 300 mutações em toda a extensão do gene, causando deficiência na produção e/ou função da proteína (referência 1; base de dados OMIM ID106100; Human Gene Mutation Database 119041; e http://hae.enzim.hu).
Angioedema hereditário por deficiência do inibidor de C1
O inibidor de C1 (C1-INH) é um potente inibidor de ativação de várias vias metabólicas, incluindo via do complemento, da coagulação e da produção de cininas (via de contato)14. A deficiência do C1-INH tem como principal consequência o aumento da ativação do sistema calicreína-cinina, com aumento da produção de bradicinina. É bem estabelecido que o aumento da bradicinina é a causa do angioedema em pacientes com AEH por deficiência do C1-INH. A bradicinina, através da ligação a receptores B2 no endotélio vascular, induz vasodilatação e aumento da permeabilidade vascular15.
O AEH tipo I representa a maioria dos casos (85%), e é resultante da deficiência quantitativa do C1-INH, enquanto que o AEH tipo II compreende 15% dos casos e é causado por uma diminuição da atividade funcional do C1-INH, com níveis séricos ou plasmáticos normais da proteína16,17. Do ponto de vista clínico, os dois tipos de AEH com deficiência do C1-INH são indistinguíveis.
A doença se caracteriza •por episódios recorrentes de angioedema que envolvem, mais frequentemente, as extremidades, face, orofaringe/laringe e os tratos gastrointestinal e genitourinário16,18. Tipicamente, os sintomas se agravam nas primeiras 24 horas e regridem lentamente nas próximas 48-72 horas. Os pacientes podem apresentar sinais prodrômicos várias horas ou até 1 dia antes dos ataques, como por exemplo, sensação estranha na pele, mal estar, fadiga, alterações de humor, e o eritema marginatum que é um rash eritematoso, não urticariforme16,17. Episódios recorrentes de edema, geralmente assimétrico, de face e extremidades são os mais frequentes, seguidos pelas crises abdominais que acometem mais de 80% dos pacientes. As crises abdominais ocorrem geralmente por edema das alças intestinais, que leva a quadros de dor abdominal intensa, acompanhada muitas vezes de náuseas, vômitos, diarreia e/ou ascite. Muitos desses episódios, quando não diagnosticados, levam à suspeita de abdome agudo e os pacientes sofrem procedimentos cirúrgicos desnecessários. O edema de vias aéreas superiores, frequentemente acometendo faringe, úvula e laringe, embora menos frequente, é o evento mais dramático para esses pacientes. Episódios de edema de laringe podem levar à asfixia e morte se não forem tratados a tempo19 (Figura 3). Mais de 50% dos pacientes com AEH sofrerão ao menos 1 episódio de edema de laringe durante a vida.
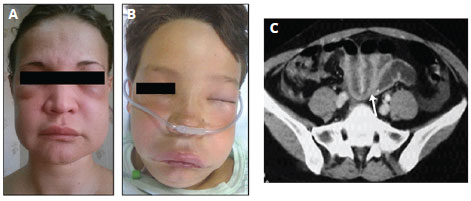
Figura 3 - Manifestações clínicas em pacientes com angioedema hereditário. A: edema subcutâneo. B: edema de vias aéreas superiores. C: edema de parede de alças intestinais (seta), visto em tomografia computadorizada de abdome
Os sintomas geralmente têm início precoce (até os 10-12 anos de idade na maioria dos casos), e é rara a ocorrência de sintomas já nos primeiros anos de vida. História familiar positiva para angioedema aumenta a suspeita clínica de AEH. No entanto, ausência de história familiar não exclui o diagnóstico, uma vez que até 25% dos pacientes podem apresentar mutações espontâneas (de novo).
Existe grande variabilidade na expressão clínica dessa doença entre os pacientes e também em um mesmo paciente ao longo dos anos. Há alguns casos assintomáticos até situações de crises extremamente graves e frequentes, trazendo importante impacto humano, social e econômico.
Na maior parte das vezes os episódios de angioedema se dão de forma espontânea, entretanto, alguns fatores têm sido associados ao desencadeamento das crises nesses pacientes, especialmente traumas físicos menores, procedimentos médicos e dentários, estresse emocional, uso de iECA, infecções, e exposição a estrógenos pelo uso de contraceptivos orais, terapia de reposição hormonal ou gravidez.
Angioedema hereditário com inibidor de C1 normal
AEH com C1-INH normal, inicialmente designado como AEH tipo III, foi clinicamente reconhecido nos anos 80 e descrito por Bork et al. em 2000. Este subtipo inclui pacientes com doença genética de característica dominante, e sintomas clínicos semelhantes aos de pacientes com AEH por deficiência do C1-INH, mas sem alterações no nível e/ou atividade funcional de C1-INH, e sem mutações no gene SERPING120.
Estudos mostram que uma proporção variável desses pacientes apresenta mutações no gene que codifica o fator XII da coagulação, F12. Duas mutações missense no exon 9 do gene F12, designadas como c.983 C > A e c.983 C > G, correspondentes a p.Thr328Lys e p.Thr328Arg foram descritas inicialmente21. Mais recentemente, duas outras mutações, uma deleção de 72 pares de base22 e uma duplicação de 18 pares de bases23, codificando uma sequência repetida de 6 aminoácidos, também no exon 9 do gene F12, foram caracterizadas em pacientes com AEH com C1-INH normal. A patogênese do AEH com C1-INH normal não é muito bem conhecida. Há evidências de que a bradicinina seja o mediador que causa o angioedema, como no AEH por deficiência do C1-INH14,24,25, entretanto, não se sabe como mutações no gene F12 levam a aumento de bradicinina. Além disso, não se conhece o defeito molecular associado a AEH com C1-INH normal sem mutações no gene F12.
As manifestações clínicas são muitos semelhantes às do AEH por deficiência de C1-INH (tipos I e II), e incluem episódios recorrentes de angioedema subcutâneo, de vias aéreas superiores e de alças intestinais, de duração entre 2 a 5 dias e alta variabilidade clínica entre os pacientes; maior gravidade dos sintomas no sexo feminino; e forte correlação com uso de estrógenos no desencadeamento ou agravamento dos sintomas. Entretanto, algumas diferenças têm sido descritas, incluindo 1) relativa maior frequência de edema cutâneo, facial, de língua, úvula e laringe e menor ocorrência de crises abdominais em relação aos pacientes com deficiência de C1-INH; 2) ausência de eritema marginatum na fase prodrômica; 3) início dos sintomas mais tardio (92% durante ou após a segunda década de vida); 4) maior proporção de pacientes apresentando episódios recorrentes numa mesma localização; 5) ocorrência de hemorragias nas áreas de angioedema, achado exclusivo desse subtipo de pacientes até o momento26. Recentemente, nosso grupo relatou pela primeira vez no Brasil a identificação da mutação c.983 C>A no gene F12, correspondente a p.Thr328Lys, em famílias de pacientes com AEH com inibidor de C1 normal. De forma interessante, maior frequência de crises de dor abdominal, e início dos sintomas numa idade mais precoce foram observados entre pacientes brasileiros com AEH com inibidor de C1 normal quando comparados àqueles de outras partes do mundo27. Stieber et al. também descreveram recentemente mutação no gene F12 em uma família brasileira de pacientes com AEH com inibidor de C1 normal28.
O registro cuidadoso da história familiar pode ser a única ferramenta para o diagnóstico do AEH com C1-INH normal. Apenas 20 a 30% desses casos apresenta mutações do gene F12, portanto, o diagnóstico de pacientes com AEH com C1-INH normal e sem mutação do fator XII é puramente baseado em evidências clínicas e história familiar característica. Os critérios diagnósticos atualmente recomendados estão descritos na Tabela 1.

ANGIOEDEMA POR DEFICIÊNCIA ADQUIRIDA DO INIBIDOR DE C1
O angioedema adquirido (AEA) ou a deficiência adquirida do inibidor de C1 é uma forma muito rara de angioedema, com prevalência estimada entre 1:100.000 a 1:500.000. Frequentemente está associado a doenças linfoproliferativas como linfoma, gamopatia monoclonal ou a doenças autoimunes, notadamente o lúpus eritematoso sistêmico (Figura 4). Esta forma não está associada a mutações no gene SERPING1, mas sim ao catabolismo aumentado do inibidor de C1. Esses pacientes costumam apresentar altos títulos de anticorpos anti-inibidor de C1, e a ligação a esses anticorpos pode desestabilizar o complexo enzima-substrato ou tornar o inibidor susceptível a clivagem proteolítica.
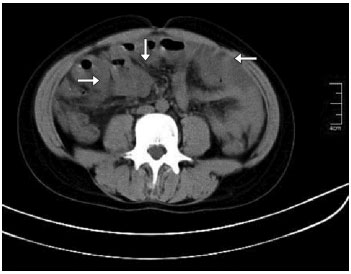
Figura 4 - Edema intenso de parede intestinal, com espessura 5 a 6 vezes maior que a normal (setas) durante ataque de dor abdominal. Paciente feminina, 17 anos de idade, com lupus eritematoso sistêmico (LES) grave, apresentou crises de angioedema subcutâneo em face e lábios, e episódios recorrentes de dor abdominal intensa, por possível deficiência adquirida do inibidor de C1. Sintomas melhoraram com o controle da atividade do LES e uso de danazol 200 mg/dia
As características clínicas dos episódios de angioedema nesses pacientes são indistinguíveis daquelas apresentadas por pacientes com AEH, incluindo as crises abdominais que são características das formas hereditárias de angioedema e da deficiência de C1-INH adquirida. Aspectos que levam à suspeita desta forma de angioedema são: 1) a idade mais tardia de início dos sintomas, já que o AEH se manifesta mais comumente na infância, enquanto AEA tende a ocorrer a partir da quarta década de vida; 2) a ausência de história familiar positiva, ao contrário da maioria dos pacientes com AEH.
A maior parte dos pacientes com deficiência adquirida de C1-INH apresenta níveis reduzidos de C1q. Laboratorialmente, essa é a uma característica que os distingue dos pacientes com AEH tipos I e II. Caso a dosagem de C1q não seja esclarecedora, o diagnóstico diferencial entre deficiência de C1-INH adquirida e deficiência de C1-INH hereditária por mutações de novo no gene SERPING1 pode ser realizado por análise genética. Todo paciente diagnosticado com AEA deve ser investigado para doenças linfoproliferativas e autoimunes, incluindo realização de hemograma com diferencial, tipagem de linfócitos (citometria de fluxo), eletroforese de proteínas, anticorpo anti-nuclear (FAN), radiografia de tórax e, possivelmente, tomografia computadorizada de tórax, abdome e pélvis, e biópsia de medula óssea, se clinicamente indicado.
ANGIOEDEMA IDIOPÁTICO NÃO HISTAMINÉRGICO
O angioedema idiopático não histaminérgico se caracteriza por ser uma forma não hereditária de angioedema em que todas as causas conhecidas de angioedema foram excluídas e os sintomas persistem apesar do tratamento contínuo com altas doses de anti-histamínicos. Não há dados que permitam estimar a frequência deste tipo de angioedema na população.
Existem algumas evidências de que a bradicinina seja o mediador envolvido na patogênese do angioedema idiopático não histaminérgico29-31. No entanto, essas evidências não são definitivas e, em pelo menos em alguns pacientes com angioedema não responsivo a anti-histamínicos, o papel de outros mediadores vasoativos como cisteinil leucotrienos, prostaglandinas e fator ativador de plaquetas deve ser considerado32.
A história clínica é o primeiro passo para o diagnóstico. Na ausência de marcadores laboratoriais, a evidência de que a histamina não é o mediador envolvido baseia-se na ausência de resposta terapêutica a altas doses (até 4 vezes a dose padrão) de anti-histamínicos de segunda geração, conforme recomendações para tratamento de urticária crônica espontânea.
Não há evidências definitivas para o tratamento das crises de angioedema idiopático não histaminérgico. A eficácia do ácido tranexâmico foi descrita em duas séries de casos33,34. Relatos de casos indicam que o icatibanto pode aliviar os sintomas nesses pacientes. Não há dados sobre a eficácia do uso de corticosteroides31. A maior parte dos dados sobre tratamento profilático nesses pacientes é sobre o uso de ácido tranexâmico. Cicardi et al.35 mostraram que uma dose de até 3 g/dia de acido tranexâmico induziu a prevenção completa (11/15) ou parcial (4/15) dos sintomas nesses pacientes. Esses resultados foram confirmados por Du-Thanh et al.34, que mostraram que até 90% dos pacientes apresentaram remissão completa ou parcial das crises de angioedema em uso de ácido tranexâmico. Drogas como ciclosporina e omalizumabe já foram testadas36, com respostas bastante variáveis, o que indica a heterogeneidade desses pacientes em relação aos mediadores envolvidos na patogênese da doença. Novos estudos buscando identificar os subgrupos de pacientes com angioedema idiopático não histaminérgico são necessários para melhor definir as estratégias de prevenção e controle das crises.
Como discutido por Riedl26, devemos considerar que é possível que esta classificação abranja um grupo heterogêneo de pacientes, podendo incluir pacientes com AEH com C1-INH normal sem história familiar (por mutações de novo) e também pacientes com urticária crônica espontânea resistente aos anti-histamínicos (lembrando que até 10% dos pacientes com urticária crônica espontânea apresentam angioedema recorrente sem urticária associada).
DIAGNÓSTICO DE ANGIOEDEMA HEREDITÁRIO E ADQUIRIDO
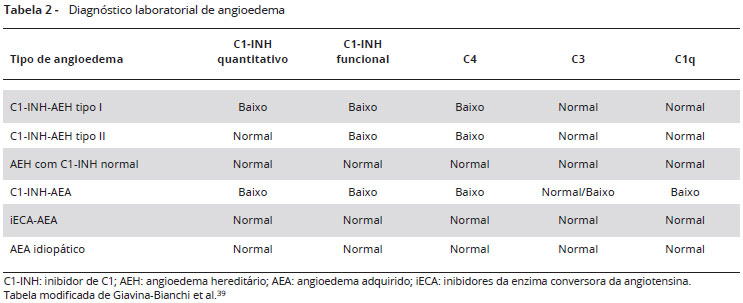
Para todas as formas de deficiência do C1-INH, a redução nos níveis de C4 é útil como bom método de screening, especialmente quando dosado na vigência de uma crise de angioedema. Níveis normais de C4 durante a crise apontam fortemente contra o diagnóstico de angioedema por deficiência de C1-INH. Caso os níveis de C4 estejam reduzidos, é necessária avaliação dos níveis séricos e atividade funcional do C1-INH. Se esses níveis estiverem baixos, recomenda-se repetir os testes para confirmação do diagnóstico37. A atividade funcional está reduzida tanto no tipo I quanto no tipo II do AEH por deficiência do C1-INH. Portanto, depois do C4, a medida da atividade do C1-INH é atualmente considerada o teste mais importante para o estabelecimento do diagnóstico38.
No AEH tipo I o nível antigênico e a atividade funcional do C1-INH estão diminuídos, enquanto que no AEH tipo II a atividade funcional está diminuída, mas o nível antigênico permanece normal. No AEH com C1-INH normal, o diagnóstico é um desafio, pois tanto o C4 quanto o nível antigênico e a atividade funcional do C1-INH estão normais. A análise genética pode ajudar nesses casos, quando é encontrada mutação no gene do FXII, entretanto nem todos os pacientes com esta condição têm mutações detectáveis nesse gene25. Os pacientes com AEA (deficiência adquirida do C1-INH) apresentam diminuição dos níveis quantitativos e/ou funcionais do C1-INH, assim como no AEH. No entanto, apenas o AEA apresenta-se com níveis de C1q diminuídos. C1q está diminuído no angioedema adquirido, e é normal nas formas hereditárias, o que pode ajudar no diagnóstico.
A Tabela 2 resume os achados laboratoriais nos diferentes tipos de AEH, bem como no angioedema adquirido e idiopático, para comparação.
Um algoritmo para auxiliar no diagnóstico das diversas formas de angioedema foi proposto por Busse e Buckland (2013)40, e está apresentado na Figura 5.
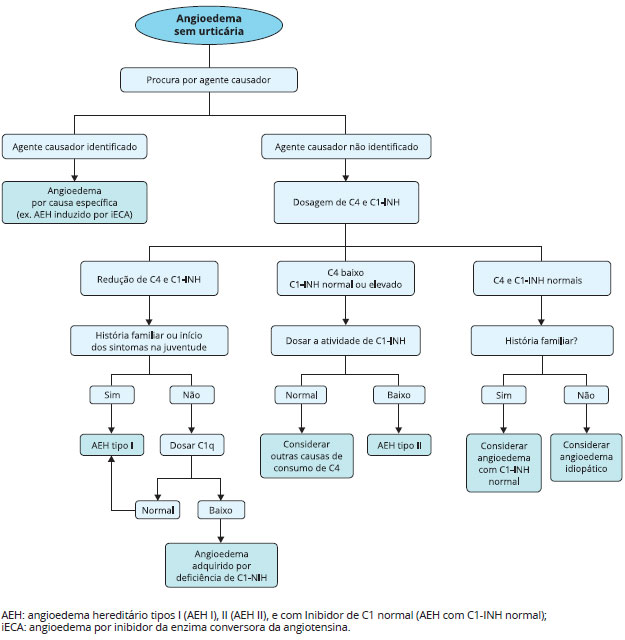
Figura 5 - Algoritmo para diagnóstico de angioedema sem urticária Modificado de Busse e Buckland (2013)40.
IMPORTÂNCIA PRÁTICA DA ANÁLISE GENÉTICA PARA PACIENTES COM ANGIOEDEMA HEREDITÁRIO E SUAS FAMÍLIAS
O diagnóstico de AEH é em geral estabelecido por sintomas clínicos característicos e testes para componentes do complemento. Entretanto, análise sistemática de mutações no gene SERPING1 tem sido realizada em várias séries de casos de AEH. Embora a determinação da mutação no gene codificador do C1-INH não seja critério essencial para o diagnóstico de AEH, a análise genética pode oferecer vários benefícios: 1) possibilitar o diagnóstico precoce de AEH, antes mesmo do aparecimento de sintomas, em membros de famílias com pacientes diagnosticados com AEH, representando valiosa ferramenta para a prevenção e tratamento precoce de crises agudas e potencialmente fatais de angioedema; 2) permitir o diagnóstico de AEH em crianças menores de um ano de idade, uma vez que nessa faixa etária os níveis séricos de C1-INH são variáveis, sendo comuns resultados falso-positivos e falso-negativos; 3) eliminar a preocupação de desenvolver a doença ou de transferir uma mutação genética para os descendentes, entre aqueles indivíduos membros de famílias de pacientes com AEH, mas que não apresentam a mutação41. Em pacientes com AEH com C1-INH normal, a análise genética é atualmente o único método que pode possibilitar o diagnóstico definitivo, uma vez que esses pacientes apresentam níveis antigênicos e funcionais de C1-INH normais, e valores de C4 normais.
Além disso, em alguns casos, a detecção de mutação no gene SERPING1 pode ser a única forma de diferenciar a forma adquirida da deficiência de C1-INH (AEA) da forma hereditária por mutação de novo, uma vez que a redução do C1q não se dá em 100% dos pacientes com AEA.
TRATAMENTO DO ANGIOEDEMA HEREDITÁRIO E DE OUTRAS FORMAS DE ANGIOEDEMA POR BRADICININA NA CRISE
O conhecimento da patogênese do AEH tem sido fundamental para o desenvolvimento de terapias específicas para o tratamento desta doença, particularmente dos ataques agudos que causam desconforto marcante e risco à vida. Essas terapias incluem a reposição do C1-INH purificado de plasma humano ou recombinante, o uso de bloqueador do receptor B2 da bradicinina (icatibanto) e de inibidor da calicreína (ecalantide), que mostram eficácia documentada no tratamento dos ataques agudos da doença37,42. Avanços no processo de preparação e purificação de C1-INH obtido a partir de plasma humano tornaram esses medicamentos mais seguros para a profilaxia a longo prazo das crises agudas43. Algumas dessas terapias têm sido também usadas em pacientes com formas adquiridas de angioedema por bradicinina.
O tratamento específico da crise deve ser instituído o mais cedo possível. O tratamento precoce é vantajoso e diminui o risco de desfechos fatais. Em alguns países a terapia domiciliar é aprovada mediante treinamento da autoadministração em todos os pacientes com AEH tipos I ou II44.
Medicações de eficácia comprovada para crises de angioedema hereditário tipos I e II
Crises de angioedema em pacientes com AEH tipos I e II devem ser tratadas com concentrado de C1-INH, icatibanto ou ecalantide, de acordo com recomendações dos consensos atuais18,39,41,42. É fortemente recomendado que os pacientes tenham disponível tratamento de uso imediato suficiente para duas crises. Se esses medicamentos não forem disponíveis, as crises devem ser tratadas com plasma tratado com detergente solvente (SDP), e quando não houver disponibilidade deste, as crises devem ser tratadas com plasma fresco congelado, preferencialmente de fonte segura. A dose recomendada de plasma fresco congelado é de 10 mL/kg. É importante enfatizar que o SDP ou o plasma fresco congelado são medicações de segunda escolha para o tratamento imediato das crises, devendo ser utilizados apenas quando o tratamento de primeira escolha (concentrado de C1-INH, icatibanto ou ecalantide) não estiver disponível. Todos os pacientes com AEH devem receber um plano de ação de manejo e de terapia imediata.
Tratamento com concentrado de C1-INH
Trata-se aqui de terapia de reposição com concentrado de C1-INH exógeno em que se elimina a causa subjacente do AEH tipos I e II, com atuação nos mesmos alvos do C1-INH endógeno.
O C1-INH derivado de plasma humano (pdC1-INH) é obtido através de procedimentos de adsorção e precipitação, purificação, pasteurização e nanofiltração. Uma unidade de pdC1-INH equivale ao conteúdo de C1-INH em 1 mL de plasma humano42. Existem três concentrados de C1-INH aprovados em diversos países para tratamento imediato de crises de AEH tipos I e II: Berinert® (CSL Behring), Cinryze® (Viropharma) e Cetor® (Sanquin). Na maioria dos países, o Berinert® é aprovado para todos os tipos de crises de AEH tipos I e II em adultos e em crianças de todas as idades (nos Estados Unidos, > 12 anos), na dose de 20U/kg de peso, por via endovenosa lenta. Cinryze® e Cetor® (1.000 U por via EV lenta) são indicados para todos os tipos de crises para idades de 12 anos e acima. Todos os produtos do pdC1-INH têm eficácia e perfil de segurança semelhantes, tendo sido relatados poucos eventos adversos. O risco de reações alérgicas é mínimo, e de transmissão de patógenos como vírus da hepatite B e C, e HIV é considerado virtualmente nulo, com o uso das técnicas modernas de purificação42,45.
Existe ainda disponível o C1-INH recombinante humano (rC1-INH), o Ruconest® (Pharming), recentemente aprovado para uso nas crises agudas de AEH pelo FDA nos Estados Unidos. Na dose de 50 U/kg de peso por via EV lenta, está recomendado para o tratamento de todas as crises de AEH tipos I e II em adultos. É produzido por glândulas mamárias de coelhas e purificado a partir do leite desses animais transgênicos. Uma unidade de rhC1-INH equivale à quantidade média de C1-INH presente em 1 mL de plasma normal fresco. É necessário que os pacientes sejam testados para a presença de IgE específica para coelho antes do início do tratamento, e posteriormente a cada ano ou a cada 10 doses da medicação, o que ocorrer primeiro. Cefaleia é um efeito adverso comum42. Ruconest® ainda não é licenciado para uso no Brasil.
Tratamento com bloqueador de receptor de bradicinina
Existe um inibidor de bradicinina disponível, o icatibanto (Firazyr®, Shire), um decapeptídeo sintético que atua impedindo de forma competitiva a ligação da bradicinina ao receptor B2 no endotélio vascular46.
O icatibanto é usado na dose de 30 mg, administrado por via subcutânea de forma lenta, e é indicado para o tratamento imediato de todos os tipos de crises de AEH tipos I e II em adultos. Na maioria dos casos, uma injeção é suficiente para tratar a crise, entretanto se não houver alívio dos sintomas ou se houver recorrência dos sintomas, uma segunda injeção pode ser aplicada após 6 horas. Se ainda não houver resposta satisfatória, uma terceira injeção de icatibanto pode ser administrada após mais 6 horas, não excedendo 3 injeções em 24 horas. Nos estudos clínicos, não foram administradas mais que 8 injeções de icatibanto por mês. Recomenda-se a aplicação na parede abdominal; é comum pacientes relatarem reações transitórias locais do tipo eritema, prurido e ardor no local da injeção, com desconforto leve.
Reações alérgicas ao icatibanto não foram relatadas. O icatibanto deve ser utilizado com cautela em condições de isquemia cardíaca aguda ou angina pectoris instável, ou nas semanas após um acidente vascular encefálico, pois pode ocorrer teoricamente deterioração da função cardíaca e redução do fluxo sanguíneo coronariano e cerebral, em razão de atenuação dos efeitos cardioprotetores e neuroprotetores mediados pela bradicinina, resultante do antagonismo do receptor B2 da bradicinina42.
Tratamento com inibidor de calicreína
O único Inibidor de calicreína disponível é o ecalantide (Kalbitor®, Dyax). É um peptídeo recombinante de 60 aminoácidos, produzido no sistema da levedura Pichia pastoris. Atua bloqueando a ação da calicreína em promover a clivagem do cininogênio de alto peso molecular, que é substrato para a produção de bradicinina, de forma que esta ação do ecalantide resulta em uma diminuição da produção de bradicinina ao final da via de ativação das cininas, inibindo a progressão do edema. O ecalantide é utilizado na dose de 30 mg em injeção subcutânea, e está indicado em todos os tipos de crises de AEH tipos I e II em pacientes maiores de 16 anos47. Anticorpos IgE e IgG contra ecalantide e contra o fungo utilizado no sistema de expressão recombinante (P. pastoris) foram demonstrados em pacientes que usaram esta medicação, e reações alérgicas têm sido relatadas.
Tratamento das crises de angioedema hereditário tipos I e II em crianças
O concentrado de pdC1-INH é o único medicamento específico aprovado para o tratamento de crises agudas de AEH tipos I e II em crianças48. O tratamento com o pdC1-INH é eficaz, bem tolerado, e tem bom perfil de segurança em crianças. Na Europa, o Berinert® na dose de 20 U/mL é aprovado para todas as idades, e nos Estados Unidos, para crianças de 12 anos e maiores. O Cinryze® (1000 U/mL) é aprovado para uso em adolescentes pós-puberais em alguns países. O C1-INH recombinante humano, o icatibanto e o ecalantide não são licenciados para uso em crianças, e a experiência na faixa etária pediátrica é muito limitada48. Quando o pdC1-INH não estiver disponível podem ser utilizados como escolhas não preferenciais o plasma tratado com detergente solvente (SDP), e se este não for disponível, o plasma fresco congelado, na dose de 10 mL/kg.
Tratamento das crises de angioedema hereditário tipos I e II durante a gravidez e lactação
Durante a gravidez e lactação, o pdC1-INH é recomendado preferencialmente para o tratamento da crise aguda de AEH, pela sua eficácia e segurança49. Não existe experiência publicada com o C1-INH recombinante humano, icatibanto ou ecalantide na gravidez ou lactação. O plasma tratado com detergente solvente (SDP) pode ser usado quando o pdC1-INH não for disponível, e o plasma fresco congelado quando o SDP não for disponível.
Tratamento das crises de angioedema hereditário com inibidor de C1 normal
De forma semelhante ao AEH tipos I e II, sintomas de AEH com C1-INH normal podem também ser debilitantes e causar risco à vida. O tratamento medicamentoso do AEH com C1-INH normal é um desafio clínico, devido à ausência de estudos controlados abordando o tratamento agudo do angioedema, uma vez que se conhece muito pouco a patogênese desta condição24. Intervenções recentes têm focalizado na abordagem terapêutica usando medicações comprovadamente eficazes no AEH por deficiência do C1-INH. Há relatos da eficácia do icatibanto no tratamento de episódios agudos de angioedema em vários pacientes com AEH com C1-INH normal. Ecalantide também mostrou eficácia durante ataque agudo em um paciente. Eficácia de terapia com pdC1-INH durante ataques agudos foi documentada em vários pacientes quando esta terapia foi tentada, embora existam também casos de insucesso26.
Tratamento da crise de angioedema por deficiência adquirida do inibidor de C1
O tratamento do AEA deve considerar a doença de base, assim como a gravidade e frequência dos episódios de angioedema. O tratamento da doença de base tem sido associado com melhora da deficiência de C1-INH e até remissão da doença, portanto deve-se investir na cura da doença de base, quando possível.
O tratamento dessa forma se assemelha ao da deficiência de C1-INH hereditária e não há resposta terapêutica ao uso de anti-histamínicos, corticosteroides ou adrenalina.
Esses pacientes requerem atenção tanto para o tratamento da crise quanto para o tratamento profilático da doença. As crises de AEA devem ser manejadas com uso de drogas reguladoras da bradicinina. Na ausência de estudos controlados para AEA, essas terapias, desenvolvidas para o tratamento do AEH, tem sido usadas off-label. Parte dos pacientes tem respondido positivamente à terapia de reposição com concentrado de C1-INH derivado de plasma, no entanto, alguns se mostram resistentes a essa terapia devido, provavelmente, ao catabolismo do C1-INH extremamente acelerado. A eficácia do icatibanto tem sido demonstrada em pequenas séries de casos, inclusive em pacientes resistentes ao C1-INH derivado de plasma50-52. Eficácia semelhante foi relatada com uso subcutâneo de ecalantide, porém reduzido número de pacientes foi avaliado até o momento47.
Alguns relatos de casos demonstram que esses pacientes podem ser tratados com rituximabe, um anticorpo monoclonal recombinante que atua no antígeno de superfície CD20 das células B. A maioria dos pacientes passou a ter episódios com menor frequência e gravidade, e alguns até entraram em remissão53-57.
Tratamento da crise de angioedema por inibidor da enzima conversora da angiotensina e outras drogas que diminuem o catabolismo da bradicinina
O tratamento consiste na descontinuação da medicação e no manejo da crise aguda na emergência. Pacientes com angioedema por iECA devem evitar qualquer outro iECA, uma vez que este efeito adverso é classe-específico. É importante considerar que a melhora do quadro pode levar cerca de 6 semanas. Até 50% dos pacientes poderão manter ocorrência de angioedema após suspensão da medicação, entretanto a frequência e gravidade das crises costumam ser bem menores58.
Pacientes com edema de vias aéreas devem ser monitorados e permanentemente avaliados, pois a intubação pode ser necessária e deve ser considerada precocemente diante de um quadro progressivo. Tratamento com anti-histamínicos e corticosteroides não se mostra eficaz no tratamento da crise e no manejo a longo prazo desses pacientes. A adrenalina pode ser considerada pela constrição temporária dos vasos sanguíneos, mas sua eficácia é reconhecidamente inferior nesses casos58.
Avanços no tratamento do AEH parecem promissores também para os pacientes com AE induzido por iECA, uma vez que compartilham da mesma via, das cininascalicreína e do mesmo mediador final, a bradicinina. Estudo recente multicêntrico, duplo cego, double-dummie, randomizado de fase 2, envolvendo 27 pacientes que procuraram Serviço de Emergência por apresentarem angioedema por iECA afetando trato aero-digestivo superior (que inclui edema de face, lábios, bochechas, língua, palato mole ou úvula, faringe e laringe), demostrou a eficácia do icatibanto. A terapia com icatibanto causou diminuição significante do tempo para resolução completa do edema, quando comparada ao tratamento combinado com prednisolona EV (500 mg) e clemastina (2 mg), com mediana de 8 horas versus 27,1 horas. Além disso, número significantemente maior de pacientes no grupo do icatibanto que no grupo da terapia com corticosteroide e anti-histamínico tiveram completa resolução do edema em 4 horas de tratamento (5/13 pacientes versus 0/14 pacientes, respectivamente)59.
Resumo das características dos medicamentos específicos utilizados nas crises agudas de AEH e outras formas de angioedema por bradicinina
Na Tabela 3 está apresentado um resumo das características dos medicamentos específicos utilizados nas crises agudas de AEH e outras formas de angioedema por bradicinina.

Tratamento não específico das crises de angioedema por bradicinina
Crises com envolvimento de vias aéreas superiores podem levar à asfixia. Embora essas crises em geral se desenvolvam ao longo de algumas horas, há relatos de instalação rápida do edema de laringe, com progressão súbita para asfixia. A presença de disfonia, disfagia e dificuldade respiratória são sinais de alerta para instituição pronta de tratamento. Em casos graves, pode ser necessária intubação endotraqueal ou traqueostomia para manutenção da via aérea. Terapia com oxigênio e monitorização por oximetria de pulso são recomendadas.
Ataques abdominais são muito dolorosos e debilitantes, podendo ser acompanhados de vômitos, diarreia e ascite. O tratamento sintomático com administração de fluidos, analgésicos, espasmolíticos, antieméticos e narcóticos podem ser necessário durante crises abdominais intensas39.
No AEH são conhecidos vários fatores desencadeantes ou agravantes de ataques agudos, que incluem: traumas físicos, cirurgias, procedimentos dentários, estresse emocional, terapia com estrógenos (contraceptivos e terapia de reposição hormonal), e uso de iECA, BRAs, e hipoglicemiantes orais do grupo das gliptinas (inibidores da DPP-4). Em crianças, infecções e trauma mecânico são fatores desencadeantes comuns. Os pacientes devem ser conscientizados sobre os desencadeantes ou agravantes de crises potencialmente relevantes, e sempre que possível estes fatores devem ser evitados. Em alguns casos como cirurgias, procedimentos dentários e situações previsíveis de estresse emocional, pode ser realizada a profilaxia a curto prazo de crises42.
PROFILAXIA DAS CRISES DE ANGIOEDEMA HEREDITÁRIO
A profilaxia das crises de AEH pode ser realizada tanto a curto prazo quanto a longo prazo. É fundamental que todo paciente que se apresenta em Pronto-Socorro com crise de AEH ou que apresente sintomas sugestivos de AEH ou angioedema por bradicinina seja encaminhado para avaliação por Alergista/Imunologista. Medicamentos como o pdC1-INH, progesterona, andrógenos atenuados particularmente o danazol, e os antifibrinolíticos ácido tranexâmico e ácido épsilon-aminocaproico têm sido utilizados de forma eficaz para prevenção de crises e de riscos associados ao AEH37.
Profilaxia de curto prazo
Existem diferentes opções eficazes para terapia profilática de curto prazo. Esta modalidade é indicada em casos onde um evento programado traga risco aumentado de crise para o paciente, como, por exemplo, procedimentos cirúrgicos ou odontológicos.
Administração de 1.000 a 2.000 U (ou 20 U/kg para crianças) de pdC1-INH é eficaz e bem tolerada na profilaxia de curto prazo. Entretanto, em muitos locais a profilaxia de curto prazo é feita com infusão de 2 U (10 mL/kg para crianças) de plasma tratado com solvente/detergente ou plasma fresco congelado, até 12 horas antes do procedimento programado. Se comparado ao plasma, o pdC1-INH garante a oferta de uma dose mais padronizada do inibidor de C1, além de serem realizados procedimentos de inativação viral mais rigorosos em sua preparação. Uma estratégia alternativa para profilaxia de curto prazo é a administração de andrógenos em altas doses (ex: 6-10 mg/kg/dia até o máximo de 200 mg 3 vezes ao dia de danazol) durante 5 a 10 dias antes e até 2 dias após o procedimento. Não existem estudos comparando a eficácia das diferentes terapias na profilaxia de curto prazo, portanto a decisão deve levar em conta o risco associado ao procedimento, os custos e a disponibilidade das terapias. Evidentemente, em procedimentos de urgência e para gestantes o uso de pdC1-INH seria preferível.
Mesmo em vigência de terapia profilática, uma dose de medicação para tratamento de crise deve estar disponível, especialmente para procedimentos dentários ou cirúrgicos que requeiram intubação.
Evitar fatores desencadeantes, tais como traumas físicos (em determinados tipos de esportes, por exemplo), estresse emocional, uso de medicamentos como iECA e estrógenos também são medidas importantes na profilaxia das crises.
Profilaxia de longo prazo
Nem todos os pacientes com AEH necessitam de profilaxia de longo prazo. A indicação deve ser individualizada, levando em conta fatores como frequência, gravidade e localização das crises, possibilidade de acesso do paciente a cuidados de emergência, disponibilidade de terapias eficazes para o tratamento das
crises e comorbidades do paciente. O objetivo da terapia profilática de longo prazo é a diminuição da frequência e da gravidade das crises de AEH60.
A reposição de pdC1-INH foi aprovada para profilaxia de longo prazo no AEH em vários países. A dose inicial é de 1.000 U a cada 3-4 dias, podendo ser ajustada conforme a resposta do paciente.
O tratamento com andrógenos atenuados por via oral também se mostra benéfico em termos da redução da frequência e gravidade das crises, no entanto, é necessário acompanhamento criterioso devido aos potenciais efeitos adversos que são, assim como a eficácia da medicação, dose-dependentes. Por isso, quando usados, deve-se buscar a mínima dose eficaz para o controle dos sintomas.
A efetividade dos agentes antifibrinolíticos (ácido tranexâmico e ácido épsilon-aminocaproico) já foi avaliada e parece ser a modalidade profilática menos efetiva63.
Uma nova droga, inibidora da calicreína administrada via oral (BCX4161), está sendo testada para o tratamento profilático do AEH. Estudo de fase 2 (OPuS-1), duplo cego, placebo controlado avaliou a segurança e a eficácia do BCX4161 como tratamento profilático para reduzir a frequência das crises em pacientes com AEH. Dados preliminares mostram que houve redução significativa do número médio de crises comparado ao placebo (p < 0,001) e melhora significante da qualidade de vida (p = 0,004) avaliada pelo AE-QoL, com perfil de segurança e tolerabilidade do BCX4161 semelhantes ao placebo6,61.
PROFILAXIA DE OUTRAS FORMAS DE ANGIOEDEMA POR BRADICININA
Andrógenos atenuados como o danazol, e antifibrinolíticos como o ácido tranexâmico, têm se mostrado efetivos na profilaxia de longo prazo em pacientes com angioedema por deficiência adquirida do C1-INH. No entanto, diferente dos pacientes com a forma hereditária, esses frequentemente respondem melhor aos antifibrinolíticos que aos andrógenos, por isso, especialistas os recomendam como droga de escolha para profilaxia de crises62. Existem relatos do uso de C1-INH derivado de plasma como terapia profilática em pequeno número de pacientes com AEA, havendo pacientes resistentes assim como quando usado para tratar as crises63.
MEDICAMENTOS PARA TRATAMENTO DE ANGIOEDEMA HEREDITÁRIO E OUTRAS FORMAS DE ANGIOEDEMA POR BRADICININA DISPONÍVEIS NO BRASIL
Icatibanto (Firazyr®, Shire) e Berinert® (CSL Behring) são licenciados e comercializados no Brasil. Ecalantide (Kalbitor®, Dyax), Cinryze® (Viropharma), Cetor® (Sanquin) e Ruconest® (Pharming) ainda não têm licenciamento para uso no Brasil. Em casos especiais pode ser feita a importação dos medicamentos ainda não licenciados, desde que os mesmos sejam aprovados em seus países de fabricação.
CONCLUSÕES
Angioedema pode representar um grande desafio diagnóstico. Cuidadosa e extensa avaliação buscando as causas conhecidas de angioedema é essencial. Quando a avaliação do complemento não é esclarecedora, é necessária investigação detalhada de aspectos da história clínica que poderão ajudar a identificar o diagnóstico etiológico. Se há história familiar de angioedema e não há medicamentos envolvidos, AEH com C1-INH normal deve ser considerado. O tratamento do AEH por deficiência de C1-INH é bem estabelecido e eficaz. Para os pacientes com diagnóstico de angioedema idiopático, uma abordagem terapêutica sistematizada poderá contribuir para esclarecer os mecanismos envolvidos no caso. Várias terapias têm sido usadas off label com sucesso variável nas diferentes formas de angioedema. No entanto, como a eficácia dessas terapias ainda não foi validada por estudos controlados, o uso dessas medicações não pode ser formalmente recomendado. Novas ferramentas diagnósticas são necessárias para esclarecer os mecanismos da forma hereditária de angioedema com C1-INH normal, bem como do angioedema idiopático e para melhor direcionar as modalidades de tratamento.
REFERÊNCIAS
1. Bork K. Angioedema. Immunol Allergy Clin North Am. 2014;34:23-31.
2. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69:602-16.
3. Rocha e Silva M, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am J Physiol. 1949;156:261-73.
4. Ferreira SH, Greene LH, Alabaster VA, Bakhle YS, Vane JR. Activity of various fractions of bradykinin potentiating factor against angiotensin I converting enzyme. Nature. 1970;225:379-80.
5. Hoover T, Lippmann M, Grouzmann E, Marceau F, Herscu P. Angiotensin converting enzyme inhibitor induced angio-oedema: a review of the pathophysiology and risk factors. Clin Exp Allergy. 2010;40:50-61.
6. Zuraw BL, Bernstein JA, Lang DM, Craig T, Dreyfus D, Hsieh F, et al. A focused parameter update: hereditary angioedema, acquired C1 inhibitor deficiency, and angiotensin-converting enzyme inhibitor-associated angioedema. J Allergy Clin Immunol. 2013;131:1491-3.
7. Bezalel S, Mahlab-Guri K, Asher I, Werner B, Sthoeger ZM. Angiotensin-converting Enzyme Inhibitor-induced Angioedema. Am J Med. 2015;128:120-25.
8. Baram M, Kommuri A, Sellers SA, Cohn JR. ACE inhibitor-induced angioedema. J Allergy Clin Immunol Pract. 2013;1:442-5.
9. Toh S, Reichman ME, Houstoun M, Ross Southworth M, Ding X, Hernandez AF, et al. Comparative risk for angioedema associated with the use of drugs that target the renin-angiotensin-aldosterone system. Arch Intern Med. 2012;172:1582-9.
10. Beavers CJ, Dunn SP, Macaulay TE. The role of angiotensin receptor blockers in patients with angiotensin-converting enzyme inhibitor-induced angioedema. Ann Pharmacother. 2011;45:520-4.
11. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, Zile MR; PARADIGM-HF Investigators and Committees. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993-1004.
12. Byrd JS, Minor DS, Elsayed R, Marshall GD. DPP-4 inhibitors and angioedema: a cause for concern? Ann Allergy Asthma Immunol. 2011;106:436-8.
13. Gosmanov AR, Fontenot EC. Sitagliptin-associated angioedema. Diabetes Care. 2012;35:e60.
14. Kaplan AP, Joseph K. Pathogenic mechanisms of bradykinin mediated diseases: dysregulation of an innate inflammatory pathway. Adv Immunol. 2014;121:41-89.
15. Kaplan AP, Joseph K. The bradykinin-forming cascade and its role in hereditary angioedema. Ann Allergy Asthma Immunol. 2010;104:193-204.
16. Longhurst H, Cicardi M. Hereditary angio-oedema. Lancet. 2012;379:474-81.
17. Ferraro M, Moreno A, Castelli E, Donadi E, Palma MS, Arcuri H, et al. A single nucleotide deletion at the C1 inhibitor gene as the cause of hereditary angioedema: insights from a Brazilian family. Allergy. 2011;66:1384-90.
18. Lang DM, Aberer W, Bernstein JA, Chng HH, Grumach AS, Hide M, et al. International consensus on hereditary and acquired angioedema. Ann Allergy Asthma Immunol. 2012;109:395-402.
19. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130:692-7.
20. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356:213-17.
21. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286-9.
22. Bork K, Wulff K, Meinke P, Wagner N, Hardt J, Witzke G. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-inhibitor. Clin Immunol. 2011;141:31-5.
23. Kiss N, Barabás E, Várnai K, Halász A, Varga LÁ, Prohászka Z, et al. Novel duplication in the F12 gene in a patient with recurrent angioedema. Clin Immunol. 2013;149:142-5.
24. Zuraw BL, Bork K, Binkley KE, Banerji A, Christiansen SC, Castaldo A, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33 Suppl 1:S145-56.
25. Bork K. Hereditary angioedema with normal C1 inhibitor. Immunol Allergy Clin North Am. 2013;33:457-70.
26. Riedl MA. Hereditary angioedema with normal C1-INH (HAE type III). J Allergy Clin Immunol Pract. 2013;1:427-32.
27. Moreno AS, Valle SO, Levy S, França AT, Serpa FS, Arcuri HA, et al. Coagulation Factor XII Gene Mutation in Brazilian Families with Hereditary Angioedema with Normal C1 Inhibitor. Int Arch Allergy Immunol. 2015 Mar 13;166(2):114-120.
28. Stieber C, Grumach AS, Cordeiro E, Constantino-Silva RN, Barth S, Hoffmann P, et al. First report of a FXII gene mutation in a Brazilian family with hereditary angioedema with normal C1 inhibitor. Br J Dermatol. 2015 Mar 27. doi: 10.1111/bjd.13791. [Epub ahead of print].
29. Cugno M, Nussberger J, Cicardi M, Agostoni A. Bradykinin and the pathophysiology of angioedema. Int Immunopharmacol. 2003;3:311-7.
30. Del Corso I, Puxeddu I, Sardano E, Geraci S, Breggia M, Rocchi V, et al. Treatment of idiopathic nonhistaminergic angioedema with bradykinin B2 receptor antagonist icatibant. Ann Allergy Asthma Immunol. 2012;108:460-1.
31. Montinaro V, Loizzo G, Zito A, Castellano G, Gesualdo L. Successful treatment of a facial attack of angioedema with icatibant in a patient with idiopathic angioedema. Am J Emerg Med. 2013;31:1295 e5-6.
32. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein J A, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69:602-16.
33. Zingale LC, Beltrami L, Zanichelli A, Maggioni L, Pappalardo E, Cicardi B, et al. Angioedema without urticaria: a large clinical survey. CMAJ. 2006;175:1065-70.
34. Du-Thanh A, Raison-Peyron N, Drouet C, Guillot B. Efficacy of tranexamic acid in sporadic idiopathic bradykinin angioedema. Allergy. 2010;65:793-5.
35. Cicardi M, Bergamaschini L, Zingale LC, Gioffré D, Agostoni A. Idiopathic nonhistaminergic angioedema. Am J Med. 1999;6:650-4.
36. Sands MF, Blume JW, Schwartz SA. Successful treatment of 3 patients with recurrent idiopathic angioedema with omalizumab. J Allergy Clin Immunol. 2007;120:979-81.
37. Cicardi M, Bork K, Caballero T, Craig T, Li HH, Longhurst H, et al. Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67:147-57.
38. Li HH, Busse P, Lumry WR, Frazer-Abel A, Levy H, Steele T, et al. Comparison of Chromogenic and ELISA Functional C1 Inhibitor Tests in Diagnosing Hereditary Angioedema. J Allergy Clin Immunol Pract. 2015;3:200-5.
39. Giavina-Bianchi P, França AT, Grumach AS, Motta AA, Fernandes FR, Campos RA, et al. Brazilian guidelines for the diagnosis and treatment of hereditary angioedema. Clinics. 2011;66:1627-36.
40. Busse PJ, Buckland MS. Non-histaminergic angioedema: focus on bradykinin-mediated angioedema. Clin Exp Allergy. 2013;43:385-94.
41. Moreno AS, Arruda LK. Tratamento da Emergência no Angioedema Hereditário. Em: Castro FFM, Watanabe AS, Silva EGM da, Arruda LK, Bittar RP. Manual de suporte avançado de vida em anafilaxia e asma. 1a ed. São Paulo: Ateneu; 2014. p. 83-91.
42. Craig T, Pursun E, Bork K, Bowen T, Boysen H, Farkas H, et al. WAO Guideline for the Management of Hereditary Angioedema. World Allergy Organ J. 2012;5:182-99.
43. Bork K. Pasteurized and nanofiltered, plasma-derived C1 esterase inhibitor concentrate for the treatment of hereditary angioedema. Immunotherapy. 2014;6:533-51.
44. Caballero T, Sala-Cunill A, Cancian M, Craig TJ, Neri S, Keith PK, et al. Current status of implementation of self-administration training in various regions of Europe, Canada and the USA in the management of hereditary angioedema. Int Arch Allergy Immunol. 2013;161:10-6.
45. Bork K, Steffensen I, Machnig T. Treatment with C1-esterase inhibitor concentrate in type I or II hereditary angioedema: A systematic literature review. Allergy Asthma Proc. 2013;34:312-27.
46. Cicardi M, Banerji A, Bracho F, Malbrán A, Rosenkranz B, Riedl M, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532-41.
47. Patel NS, Fung SM, Zanichelli A, Cicardi M, Cohn JR. Ecallantide for treatment of acute attacks of acquired C1 esterase inhibitor deficiency. Allergy Asthma Proc. 2013;34:72-7.
48. Farkas H, Csuka D, Zotter Z, Szabó E, Czaller I, Varga L, et al. Treatment of attacks with plasma-derived C1-inhibitor concentrate in pediatric hereditary angioedema patients. J Allergy Clin Immunol. 2013;131:909-11.
49. Caballero T, Farkas H, Bouillet L, Bowen T, Bork K, Bygum A, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129(2):308-20.
50. Bright P, Dempster J, Longhurst H. Successful treatment of acquired C1 inhibitor deficiency with icatibant. Clin Exp Dermatol. 2010;35:553-4.
51. Weller K, Magerl M, Maurer M. Successful treatment of an acute attack of acquired angioedema with the bradykinin-B2-receptor antagonist icatibant. J Eur Acad Dermatol Venereol. 2011;25:119-20.
52. Zanichelli A, Bova M, Coerezza A, Petraroli A, Triggiani M, Cicardi M. Icatibant treatment for acquired C1-inhibitor deficiency: a real-world observational study. Allergy. 2012;67:1074-7.
53. Branellec A, Bouillet L, Javaud N, Mekinian A, Boccon-Gibod I, Blanchard-Delaunay C, et al. Acquired C1-inhibitor deficiency: 7 patients treated with rituximab. J Clin Immunol. 2012;32:936-41.
54. Levi M, Hack CE, van Oers MH. Rituximab-induced elimination of acquired angioedema due to C1-inhibitor deficiency. Am J Med. 2006;119:e3-5.
55. Hassan A, Amarger S, Tridon A, Ponard D, Souteyrand P, D'Incan M. Acquired angioedema responding to rituximab. Acta Derm Venereol. 2011;91:733-4.
56. Lam DH, Levy NB, Nickerson JM, Gruenberg DA, Lansigan F. Acquired angioedema and marginal zone lymphoma. J Clin Oncol. 2012;30:e151-3.
57. Ziakas P, Giannouli S, Psimenou E, Evangelia K, Tzioufas A, Voulgarelis M. Acquired angioedema: a new target for rituximab? Haematologica. 2004;89:ELT13.
58. Beltrami L, Zanichelli A, Zingale L, Vacchini R, Carugo S, Cicardi M. Long-term follow-up of 111 patients with angiotensin-converting enzyme inhibitor-related angioedema. J Hypertens. 2011;29:2273-7.
59. Bas M, Greve J, Stelter K, Havel M, Strassen U, Rotter N, et al. A randomized trial of icatibant in ACE-inhibitor-induced angioedema. New Engl J Med. 2015;29;372:418-25.
60. Zuraw BL, Banerji A, Bernstein JA, Busse PJ, Christiansen SC, Davis-Lorton M, et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J Allergy Clin Immunol Pract. 2013;1:458-67.
61. Aygören-Pürsün E, Magerl M, Graff J, Martinez-Saguer I, Kreuz W, Longhurst H, et al. Efficacy correlates with plasma levels in Opus-1, a proof-of concept study of oral kallikrein inhibitor BCX4161 as a prophylaxis against attacks of hereditary angioedema (HAE). J Allergy Clin Immunol. 2015;135:AB192.
62. Bork K. Current drugs in early development for hereditary angioedema: potential for effective treatment. Expert Opin Investig Drugs. 2014;23:887-91.
63. Zingale LC, Castelli R, Zanichelli A, Cicardi M. Acquired deficiency of the inhibitor of the first complement component: presentation, diagnosis, course, and conventional management. Immunol Allergy Clin North Am. 2006;26:669-90.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888