Número Atual: Julho-Setembro 2023 - Volume 7 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicações Clínicas e Experimentais
Hipogamaglobulinemia e rituximabe: relato de casos
Hypogammaglobulinemia and rituximab: case reports
Márcio Niemeyer Souza1; Luciana Costa Ribeiro2; Raisa Gusso Ulaf2; Thyago Alves Nunes2; Eli Mansour2
1. Goiânia, GO, Brasil
2. UNICAMP, Serviço de Imunologia e Alergia - Campinas, SP, Brasil
Endereço para correspondência:
Márcio Niemeyer Souza
E-mail: marcioniemeyer@gamil.com
Submetido em: 07/08/2023
Aceito em: 27/08/2023.
RESUMO
As imunodeficiências secundárias podem ser uma consequência de neoplasias, infecções ou tratamentos imunossupressores. O rituximabe (RTX) é um anticorpo anti-CD20 que depleta os linfócitos B e pode induzir uma hipogamaglobulinemia sintomática. Aqui, relatamos três casos de hipogamaglobulinemia sintomática associada ao uso de RTX. Na primeira paciente com artrite reumatoide, o RTX induziu a baixos níveis de imunoglobulinas associadas a infecções de vias aéreas de repetição. Após a suspensão do RTX, houve normalização da resposta imune humoral. Os outros dois casos, com o uso de RTX para tratamento de linfoma não-Hodgkin e lúpus eritematoso sistêmico, respectivamente, as pacientes evoluíram com hipogamaglobulinemia secundária persistente, com necessidade de reposição de imunoglobulina por vários anos. Pacientes tratados com RTX podem apresentar, após a sua suspensão, uma recuperação rápida da função humoral ou permanecerem com baixos níveis séricos de imunoglobulinas por longos períodos. Com o crescente uso dos tratamentos direcionados para componentes do sistema imunológico, um alto grau de suspeição clínica para o aparecimento de imunodeficiências secundárias pode minimizar a morbimortalidade associada a estes tratamentos.
Descritores: Rituximab, artrite reumatoide, antibioticoprofilaxia.
Introdução
As Imunodeficiências, condições que afetam o funcionamento correto do sistema imunológico, podem ser classificadas em primárias (IDP), recentemente renomeadas em erros inatos da imunidade, ou secundárias. As imunodeficiências secundárias (IDS) podem ocorrer como consequência de doença subjacente, como neoplasia ou infecção, ou tratamento imunossupressor. As IDS são mais prevalentes que as IDP e frequentemente não são reconhecidas pelos clínicos1.
As desordens de linfócitos B são os subtipos de imunodeficiências mais comuns, responsáveis por aproximadamente 50% dos diagnósticos de IDP. Caracterizam-se por um aumento da suscetibilidade a infecções do trato respiratório por bactérias, particularmente pelo Streptococcus pneumoniae e Haemophilus influenzae. Os sintomas apresentam-se com infecções sino-pulmonares recorrentes como otite média, sinusite e pneumonia. Diarreia, fadiga, manifestações autoimunes, particularmente citopenias, e perda auditiva também são comuns. Pacientes com imunodeficiência humoral têm níveis séricos de imunoglobulinas diminuídos ou ausentes, mas podem apresentar níveis normais ou aumentados, porém com função anormal2-4.
O rituximabe (RTX), um anticorpo monoclonal anti-CD20, foi o primeiro anticorpo aprovado inicialmente para o tratamento de linfoma não-Hodgkin (LNH) de células B CD20+ folicular ou de baixo grau recidivante ou refratário, em 1997. Seu uso foi então estendido para o tratamento de outras doenças malignas de células B e para doenças não-malignas, como artrite reumatoide (AR). A depleção dos linfócitos B em pacientes com artrite reumatoide pelo RTX, tanto como monoterapia como associado à ciclofosfamida e/ou corticosteroides, é um arsenal terapêutico desde 19983.
A ligação do RTX ao CD20 pode resultar na apoptose dos linfócitos B por mecanismos ainda não completamente esclarecidos. A ligação do RTX pode, também, ativar a via clássica do complemento, resultando na formação do complexo de ataque à membrana (MAC) e lise celular. Adicionalmente, recruta as células natural killer através de sua ligação ao FcϒRII levando à citotoxicidade celular mediada por anticorpo (ADCC) e por último, os macrófagos fagocitam a célula-alvo através do reconhecimento do CD20 ligado ao RTX3. Após o tratamento com RTX, em torno de 38,5% dos pacientes têm baixos níveis de IgG, e infecções sino-pulmonares foram vistas, em média, em 6,6%. A reposição de imunoglobulinas parece reduzir a frequência das infecções4. A depleção de imunoglobulina M (IgM) após o uso de RTX é mais frequente e prolongada que a de IgG, mas clinicamente menos significativa5. Aqui apresentamos e discutimos 3 casos de hipogamaglobulinemia associada ao uso de RTX, no contexto de linfoma ou autoimunidade.
Apresentação dos casos
Caso 1
V.F.P.D., feminina, 47 anos de idade e 11 anos de diagnóstico de AR. Após vários esquemas terapêuticos sem boa resposta, o RTX foi iniciado com controle sintomático da doença. Seis meses após a introdução do RTX, a paciente iniciou um quadro de sinusites agudas recorrentes, num total de 5 episódios no último ano, com posterior cronificação e pneumonias de repetição com 5 episódios de pneumonia no mesmo período (Tabela 1). No último mês relata dois episódios de internação hospitalar com o uso de antibióticos de largo espectro por via intravenosa (meropenem) para tratamento de pneumonia. As tomografias computadorizadas dos seios paranasais (Figura 1A e 1B) e de tórax (Figura 2A e 2B), feitas fora da crise infecciosa, mostram acometimento pansinusal e bronquiectasias, respectivamente. Não apresentava sintomas respiratórios baixos, com ausculta pulmonar sem alterações ao exame físico, mas com moderada quantidade de rinorreia mucopurulenta bilateral na consulta de avaliação inicial.
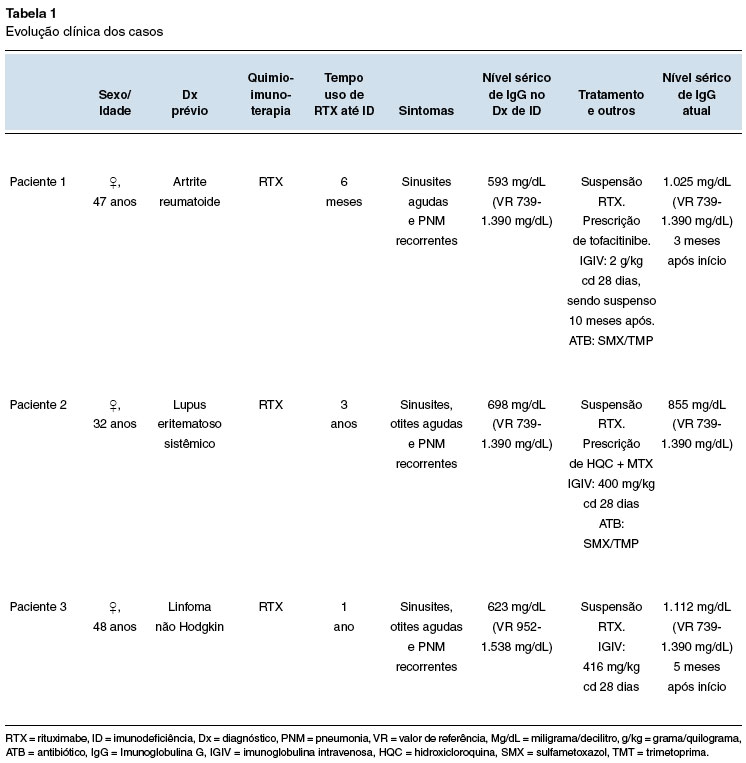
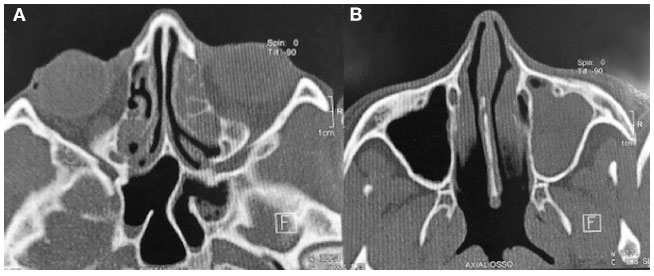
Figura 1
TC de seios paranasais. Corte axial com velamento difuso de células etmoidais, do seio maxilar direito e presença de secreção em seios esfenoidais
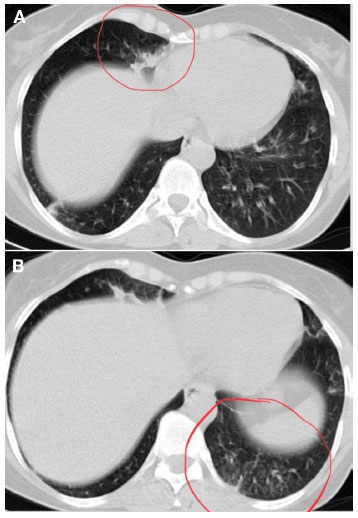
Figura 2
TC de tórax mostrando bronquiectasia de língula e estase de secreção mucoide em bronquíolos periféricos
A história familiar é de uma tia materna com diagnóstico de AR. Os exames laboratoriais mostram (valores de referência do laboratório para a idade): IgM: 25 mg/dL (VR: 33-293); IgG: 593 mg/dL (VR: 739-1390); IgA: 129 mg/dL (VR: 65-421); IgE: 46,2 kU/L (<140 kU/L); a eletroforese de proteínas séricas: Gamaglobulina: 8,8%-0,55 g/dL (10,3 a 18,2% de 0,74 a 1,75 g/dL) (Tabela 2) e demais exames de rotina estavam normais. A imunofenotipagem de subpopulações de linfócitos com ausência de linfócitos B (CD19), < 1% dos linfócitos do sangue periférico, e demais células normais (vide Tabela 3). Feito o diagnóstico de hipogamaglobulinemia secundária ao uso de RTX, foi iniciada antibioticoterapia profilática com sulfametoxazol/trimetoprim (SMX/TMP) na dose terapêutica de 800/160 mg duas vezes ao dia e indicada reposição com imunoglobulina humana intravenosa (IVIG). A IVIG foi prescrita na dose imunomoduladora, pelo reumatologista assistente, de 2 g/kg de peso para a primeira aplicação. O reumatologista preferiu suspender o RTX e prescrever tofacitinibe. Após a terapia com IVIG iniciada 1 mês após a consulta inicial e troca do RTX houve normalização da IgG sérica total (1.025 mg/dL) cerca de 3 meses depois da primeira consulta. Desde então a paciente não apresentou mais quadros infecciosos em seguimento há mais de 10 meses. Mantém-se controlada da AR, a antibioticoprofilaxia com SMX/TMP foi suspensa após a normalização dos níveis séricos de IgG (Tabela 2). Também houve a normalização do quadro radiológico sinusal e melhora do pulmonar (Figura 3A e 3B).
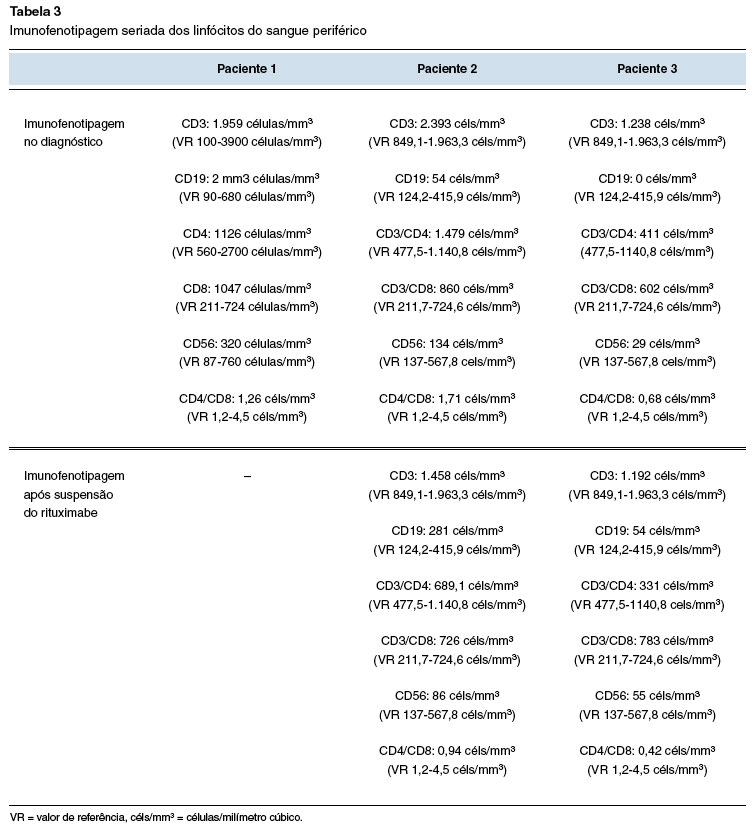
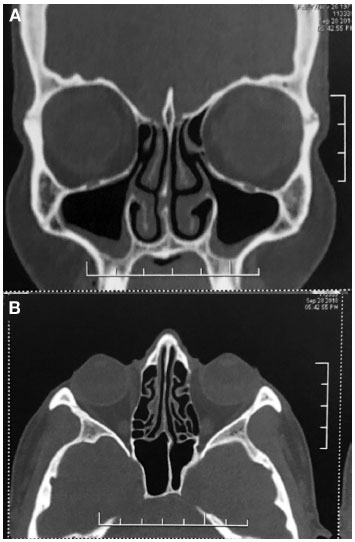
Figura 3
TC de seios paranasais - realizada 6 meses após o primeiro exame (Figura 1) mostrando normalização da doença sinusal. Permanece com leve espessamento mucoso inespecífico em assoalho de seios maxilares
Caso 2
J.T.V., feminina e 48 anos de idade, com quadro de LNH tratada em 2012 com quimioterapia e RTX. Cerca de um ano após uso do RTX, iniciou as infecções de repetição com diversas pneumonias, otites e sinusites, e necessidade de antibioticoterapia (Tabela 1). A tomografia de seios da face mostrava espessamento pansinusal (Figura 4A e 4B). Em dezembro de 2018 foi encaminhada ao serviço de imunologia, quando recebeu o diagnóstico de hipogamaglobulinemia (Tabela 2).
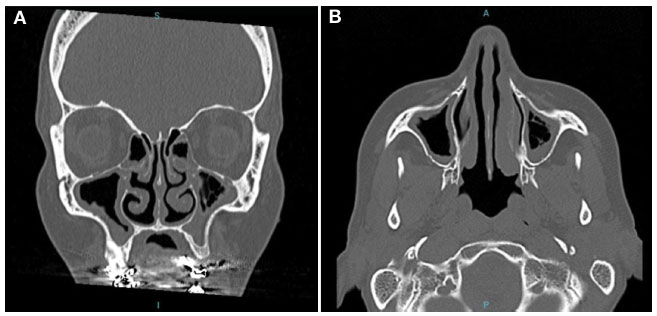
Figura 4
TC de seios da face de setembro de 2014 (cortes coronal e axial) demonstrando espessamento mucoso dos seios maxilares bilateralmente, de algumas células etmoidais
Os exames laboratoriais de dezembro de 2018 mostravam IgA: < 6,4 mg/dL (VR 153-359 mg/dL); IgE: < 4,3 mg/dL (VR < 100 mg/dL); IgG: 623 mg/dL (VR 952-1538 mg/dL); IgM: 17,5 mg/dL (VR 73-171 mg/dL). A imunofenotipagem de subpopulações de linfócitos mostrava ausência de linfócitos B (Tabela 3). A sorologia para pneumococos de janeiro de 2019: sorotipo 4: inferior 0,5; sorotipo 6b: inferior a 0,5; sorotipo 9v: inferior a 0,5; sorotipo 14: 3,4; sorotipo 18c: inferior a 0,5; sorotipo 19f: 1,2; sorotipo 23f: inferior a 0,5.
As dosagens de imunoglobulinas foram repetidas em fevereiro de 2019 com IgG: 623 mg/dL; IgE: < 4,3 mg/dL; IgM: < 17,5 mg/dL; IgA: < 6,4 mg/dL e a imunofenotipagem confirmando ausência de células B (Tabela 3). Devido à hipogamaglobulinemia sintomática e à ausência das células B, a imunoglobulina humana intravenosa (IVIG) foi iniciada na dose de 25 g (416 mg/kg) a cada 28 dias em maio de 2019. Após introdução da IVIG, a paciente não apresentou mais quadros infecciosos nem necessidade de antibioticoterapia. Uma nova dosagem de imunoglobulinas realizada em julho de 2019 e, portanto, dois meses após início de uso da IVIG: IgE: < 2 mg/dL; IgA: < 10 mg/dL; IgG: 1112 mg/dL; IgM: < 20 mg/dL. A deficiência dos linfócitos B no sangue periférico continua evidente numa nova imunofenotipagem (Tabela 3) de janeiro de 2020, e por isso a reposição de imunoglobulinas com IVIG foi mantida na mesma dose, na dependência de futuras reavaliações.
Caso 3
P.R.V.F., 32 anos de idade, feminina, e diagnóstico inicial de policondrite recidivante há cerca de 10 anos tratada inicialmente com metotrexato e prednisona. Três anos após, por apresentar úlceras orais e esofágicas, policondrite de pequenas e grandes articulações e eritema malar associados a alterações laboratoriais como aumento de proteína C-reativa, positividade do fator antinuclear (FAN) e anticorpos anti-DNA, foi então diagnosticada com lúpus eritematoso sistêmico. Após falha terapêutica com tocilizumabe, o RTX foi introduzido com melhora sintomática significativa três meses após.
A paciente então passou a apresentar infecções respiratórias graves recorrentes, cerca de 3 anos após o início do RTX, em uma delas internada em unidade de terapia intensiva com quadro de sepse secundária à sinusite. Nos 4 anos subsequentes, apresentou um total de 25 episódios de sinusite e 2 pneumonias (Tabela 1). Os exames laboratoriais realizados cerca de 3 anos após o início do RTX mostram depleção de linfócitos B, bem como hipogamaglobulinemia (Tabelas 2 e 3): IgG: 698 mg/dL (VR: 739-1390), IgA: 57 mg/dL (VR: 65-421); IgM: 22 mg/dL (VR: 33-293; linfócitos B CD20: 54 cél/mm3 (90-680 mm3). Optou-se então pela suspensão do RTX e troca pela hidroxicloroquina associada ao metotrexato. Após 2 anos de infecções sustentadas foi iniciada antibioticoprofilaxia com SMX/TMP, o que não impediu novas infecções. Sem resposta à antibioticoprofilaxia, tratamento escolhido foi reposição de IGIV na dose de 400 mg/kg a cada 28 dias. Seis meses após a primeira aplicação de IGIV foi tentada uma suspensão, mas os níveis de imunoglobulinas continuaram em queda progressiva, apesar da normalização dos linfócitos B. A reposição de IGIV na mesma dose foi reiniciada e o diagnóstico de imunodeficiência comum variável foi suspeito.
Discussão
Neste relato de casos avaliamos três pacientes do sexo feminino na faixa etária da quarta e quinta décadas de vida, das quais duas apresentavam doenças autoimunes, AR (caso 1) e lúpus eritematoso sistêmico (caso 3), e o caso 2, uma neoplasia (LNH).
A três pacientes utilizaram em algum momento de seu esquema terapêutico, o RTX, um anticorpo monoclonal quimérico anti-CD20. Este medicamento depleta os linfócitos B, mas somente 38,5% dos pacientes apresentam hipogamaglobulinemia e apenas cerca de 6,6% hipogamaglobulinemia sintomática5, que se expressa clinicamente como infecções sino-pulmonares de repetição. Como se tratam de infecções corriqueiras, embora se apresentem de forma recorrente, causadas por germes típicos, estes pacientes podem ter o diagnóstico de hipogamaglobulinemia sintomática secundária feito de forma tardia, ou seja, quando já apresentam sequelas, como bronquiectasias, tal como ocorreu no caso 1. De tal forma a se evitar tais complicações, é necessário um alto grau de suspeição clínica. A hipogamaglobulinemia sintomática foi apresentada pelas três pacientes descritas neste relato. Optou-se após o diagnóstico, em fazer a reposição de IGIV, no primeiro caso, inicialmente em dose imunomoduladora, e nas demais, repositória.
No primeiro caso, para evitar o tratamento com dois medicamentos de alto custo, o reumatologista optou pela troca do RTX por outro esquema terapêutico com vistas ao controle da doença de base (AR). Ainda neste primeiro caso, os níveis de IgG normalizaram-se após a suspensão do RTX e troca do esquema terapêutico. A paciente não retornou a apresentar novas infecções até o término do acompanhamento. Esta paciente era acompanhada em consultório particular e morava em cidade distante. Além disto, em dado momento perdeu seu seguro saúde, o que tornou impossível realizar exames laboratoriais tal como ocorreu nos outros dois casos, que eram acompanhadas em unidade pública de saúde.
No terceiro caso, após a suspensão do RTX, como não houve normalização das taxas das imunoglobulinas e a paciente apresentava doença autoimune e por isso, o diagnóstico de imunodeficiência comum variável foi aventado. A imunodeficiência comum variável é a imunodeficiência primária mais comum que se expressa nos adultos. A disfunção dos linfócitos B leva a baixos níveis séricos de IgG e usualmente de IgA e/ou de IgM. Como consequência, os pacientes podem desenvolver, frequentemente numa etapa mais tardia da vida, e infecções bacterianas recorrentes das vias aéreas. Outras desordens clínicas foram largamente descritas como enteropatia crônica, doenças linfoproliferativas malignas e benignas, doenças granulomatosas e desordens autoimunes6-8. Assim, as infecções respiratórias recorrentes podem ser devido à imunodeficiência comum variável e não ao uso do RTX, mas a paciente persiste em reposição de IGIV e não mais apresentou infecções respiratórias recorrentes. Embora as infecções recorrentes sejam a principal forma de apresentação das imunodeficiências primárias, a autoimunidade está presente em um grande número de IDPs devido à perda da autotolerância9.
No segundo caso, a paciente continua a reposição com IGIV, vários anos após a suspensão do RTX para o tratamento de linfoma não-Hodgkin, por manter a hipogamaglobulinemia. Barmettler e cols. dividem os pacientes com hipogamaglobulinemia após tratamento com RTX em dois subgrupos: um com recuperação normal de imunoglobulinas e linfócitos B, e um outro com hipogamaglobulinemia sintomática de longa duração. Em pacientes com hipogamaglobulinemia persistente há um risco aumentado de infecções devido à disfunção/deficiência humoral, e estes pacientes podem se beneficiar da reposição de IGIV6. Provavelmente esta paciente pertence ao segundo grupo, o da hipogamaglobulinemia sustentada.
O caso 1 pertence provavelmente ao primeiro subgrupo supracitado, com recuperação rápida dos níveis de imunoglobulinas e linfócitos B; e o caso 2, possivelmente pertencente ao segundo subgrupo, o dos pacientes que persistem com hipogamaglobulinemia por períodos longos. Este último subgrupo tem uma média de duração de terapia de reposição com IGIV de 83 meses, após a suspensão do RTX, comparados com média de 24 meses do primeiro subgrupo6, por isso que no caso 3, mesmo com diminuição de outras classes de imunoglobulinas, o diagnóstico de IDCV ainda não pôde ser concluído. Em pacientes com hipogamaglobulinemia sustentada pode ser desafiador distingui-los das imunodeficiências primárias, especialmente da imunodeficiência comum variável10.
Conclusões
Nos três casos expostos, as pacientes desenvolveram um quadro de imunodeficiência humoral que, se não diagnosticado a tempo, poderia levar a repercussões clínicas drásticas. Em dois dos casos, o caso 1 e o caso 3, houve um atraso significativo entre o início do quadro sindrômico de imunodeficiência humoral e o seu diagnóstico. No caso 1, as sequelas pulmonares se mostraram irreversíveis mesmo após o tratamento adequado. Com o desenvolvimento de novos anticorpos monoclonais para o tratamento das mais variadas doenças e principalmente em pacientes em uso de anticorpos antilinfócitos B, é necessário um alto grau de suspeição clínica para o aparecimento de hipogamaglobulinemia secundária sintomática e início de antibioticoprofilaxia e, caso necessário, de reposição de imunoglobulinas.
Referências
1. Kaplan B, Bonagura VR. Secondary hypogammaglobulinemia: an increasingly recognized complication of treatment with immunomodulators and after solid organ transplantation. Immunol Allergy Clin North Am. 2019;39(1):31-47.
2. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016:4(1):38-59.
3. Tangye SG, Waleed Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on classification from International Union of Immunological Societies expert committee. J Clin Immunol. 2020;40(1):24-64.
4. Bousfiha A, Jeddane L, Picard C, Al-Herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. 2020;40(1):66-81.
5. Pierpont TM, Limper CB, Richards K. Past, present and future of rituximab - the world's first monoclonal antibody therapy. Front Oncol. 2018;8:163.
6. Barmettler S, Ong MS, Farmer JR, Choi H, Walter J. Association of immunoglobulin levels, Infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open. 2018:1(7):e184169.
7. Boileau J, Mouillot G, Gérard L, Carmagnat M, Rabian C, Oksenhendler E, et al. Autoimmunity in common variable immunodeficiency: Correlation with lymphocyte phenotype in French DEFI study. J Autoimmun. 2011;36(1):25-32.
8. Agarwal S, Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency. Ann Allergy Asthma Immunol. 2019;123(5):454-60.
9. Hoyos-Bachiglu R, Chou Janet. Autoimmunity and Immunodeficiency. Curr Opin Rheumatol. 2020,32:168-74.
10. Barmettler S, Price C. Continuing IgG replacement therapy for hypogammaglobulinemia after rituximab-for how long? J Allergy Clin Immunol. 2015;136(5):1407-9.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888