Número Atual: Julho-Setembro 2023 - Volume 7 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo Original
Doença granulomatosa crônica: características clínicas, seguimento e terapêutica de cinco pacientes pediátricos
Chronic granulomatous disease: clinical characteristics, follow-up, and therapy of 5 pediatric patients
Karina Mescouto de Melo1; Ludmila Gonçalves Ribeiro1; Claudia França Cavalcante Valente1; Carmem Maria Sales Bonfim2; Antonio Condino-Neto3; Shirley Claudino Pereira Couto4; Maria Imaculada Muniz-Junqueira4; Simone Castro Resende Franco5; Thalita Dias6; Fabíola Scancetti Tavares1
1. Hospital da Criança de Brasília José Alencar, Serviço de Alergia e Imunologia - Brasília, DF, Brasil
2. Hospital de Clínicas da Universidade Federal do Paraná, Serviço de transplante de Medula Óssea - Curitiba, PR, Brasil
3. Universidade de São Paulo, Departamento de Imunologia, Instituto de Ciências Biomédicas - São Paulo, SP, Brasil
4. Universidade de Brasília, Laboratório de Imunologia Celular, Faculdade de Medicina - Brasília, DF, Brasil
5. Hospital da Criança de Brasília José Alencar, Serviço de Onco-hematologia - Brasília, DF, Brasil
6. Hospital de Base do Distrito Federal Brasília, Serviço de Alergia e Imunologia - Brasília, DF, Brasil
Endereço para correspondência:
Karina Mescouto de Melo
E-mail: karinamescouto@gmail.com
Submetido em: 02/08/2023
Aceito em: 24/08/2023.
RESUMO
INTRODUÇÃO: A doença granulomatosa crônica (DGC) é caracterizada por um defeito na capacidade microbicida das células fagocíticas (monócitos e neutrófilos), com alta mortalidade se não diagnosticada precocemente. Os pacientes apresentam infecções recorrentes ou graves, suscetibilidade a granulomas em órgãos profundos, doenças autoimunes e doença inflamatória intestinal.
OBJETIVO E MÉTODO: Relato de aspectos clínicos e do tratamento de cinco pacientes com doença granulomatosa crônica.
RESULTADOS: Cinco pacientes, três meninos, medianas de idade no início dos sintomas e diagnóstico de 8 meses e 48 meses, respectivamente, foram estudados por um período de 10 anos. Pneumonia (5/5) e doença micobacteriana (3/5) foram as manifestações iniciais mais comuns. Alterações pulmonares foram observadas em todos os casos. Mutações nos genes CYBB e NCF1 foram identificadas em três casos. Antibioticoprofilaxia foi instituída em todos os pacientes e três foram submetidos ao transplante de células tronco-hematopoiéticas (TCH), aos 7, 18 e 19 anos e com sobrevida atual entre 4 a 5 anos.
CONCLUSÃO: O monitoramento cuidadoso de infecções graves com tratamento imediato foi crucial para a sobrevivência. O TCH, mesmo ao final da adolescência, promoveu a cura da DGC em três pacientes.
Descritores: Doença granulomatosa crônica, fagócitos, NADPH oxidases, transplante de medula óssea
Introdução
A doença granulomatosa crônica (DGC) é uma imunodeficiência primária (IDP) ou erro inato da imunidade (EII) grave, caracterizada por defeito na função microbicida das células fagocíticas (neutrófilos e monócitos)1,2. A DGC tem incidência estimada em 1:200.00 nascidos vivos2. O início dos sintomas ocorre nos primeiros anos de vida, com infecções graves em pele, trato respiratório, linfonodo, fígado e ossos, além de suscetibilidade a granulomas em órgãos profundos e doenças autoimunes2-4.
A DGC é causada por mutações em um dos genes que codificam as proteínas do complexo enzimático nicotinamida adenina dinucleotídeo (NADPH) oxidase nas células fagocíticas. O gene CYBB, que codifica a proteína gp91phox, causa a DGC ligada ao cromossomo X e representa aproximadamente 70% dos casos1,2. Há ainda as formas autossômicas recessivas, decorrentes de mutações nos genes NCF1, NCF2, CYBA, NCF4 e CYBC11,3.
Os critérios diagnósticos incluem: (I) critérios clínicos: infecção grave por bactérias e/ou fungos (abscessos, osteomielite, linfadenite), pneumonia recorrente, linfadenopatia e/ou hepatomegalia e/ou esplenomegalia, granulomas obstrutivos/difusos (trato gastrointestinal ou urogenital), e manifestações inflamatórias crônicas (colite, abscesso hepático e formação de fístula)4; (II) critérios laboratoriais: (a) alteração em dois testes de oxidação de células fagocíticas, pelo nitroblue-tetrazolium (NBT) por microscopia ótica ou di-hidrorodamina (DHR) por citometria de fluxo2,4, ou (b) identificação de mutação em um dos genes associados à DGC1-3. Para o diagnóstico definitivo é necessário pelo menos um critério clínico e um critério laboratorial4.
A profilaxia com sulfametoxazol-trimetoprim (5 mg/kg/dia de trimetoprim) e itraconazol 100 mg ao dia para pacientes < 13 anos ou < 50 kg; 200 mg diariamente para aqueles ≥ 13 anos ou ≥ 50 kg, é o tratamento convencional para DGC2,3. No entanto, o transplante de células hematopoiéticas (TCH) é o único tratamento curativo2,3. Há poucos dados disponíveis sobre TCH na DGC em nosso país5,6. Neste estudo descrevemos o seguimento clínico de dez anos de cinco casos pediátricos, destacando o TCH como terapêutica bem-sucedida em três casos.
Descrição
Cinco pacientes com DGC acompanhados em serviço de imunologia de hospital pediátrico terciário do Distrito Federal foram estudados. Os dados foram coletados de prontuários dos pacientes e compreende o período de 2011 a 2021. Os exames confirmatórios foram realizados em cooperação com os laboratórios de Imunologia Celular da Universidade Brasília (UNB), no caso do teste NBT; e laboratório de Imunologia Humana da Universidade de São Paulo, para os testes de DHR e análise genética. O estudo foi aprovado pelo Comitê de Ética em Pesquisa /Plataforma Brasil.
Características clínicas
Três pacientes são meninos (60%), com medianas de idade no início dos sintomas e diagnóstico de 8 e 48 meses, respectivamente. Pneumonia grave foi a principal manifestação clínica inicial da doença, observada em todos os casos. Uma criança (P-1) tinha histórico de óbitos precoces na família, dois tios maternos, cinco primos maternos e um irmão por pneumonia (Tabela 1). Uma paciente (P-3) foi encaminhada do serviço de onco-hematologia devido a quadro de anemia hemolítica autoimune associada a pneumonia grave.
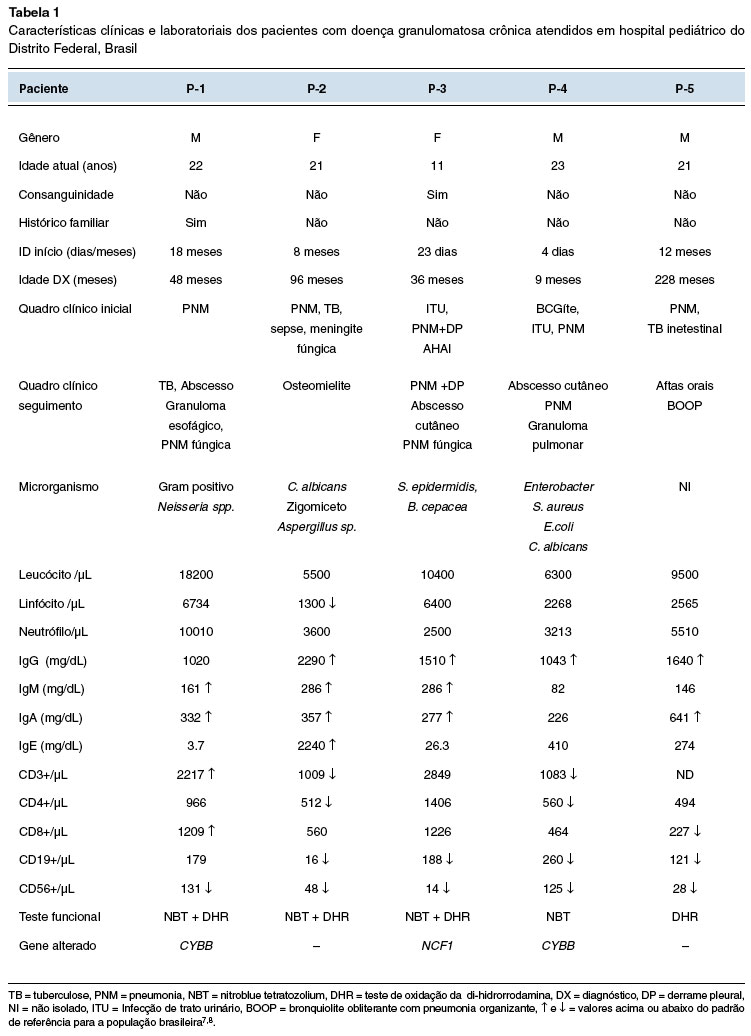
Todos os pacientes necessitaram de internação hospitalar devido a quadro de infecção antes do diagnóstico. Todos os pacientes estão vivos.
Exames complementares laboratoriais
Na avaliação inicial do sistema imune, foi observada hipergamaglobulinemia7 em 4/5 pacientes. Houve aumento no número de linfócitos T CD3+ em dois pacientes (P-1 e P-3) e todos apresentaram diminuição do número absoluto de células natural killer (NK)8 (Tabela 1). Os testes para avaliação da capacidade microbicida dos neutrófilos (NBT ou DHR) e/ou sequenciamento genético foram feitos para a confirmação da doença em todos os pacientes (Tabela 1). Dois pacientes apresentaram as seguintes mutações no gene CYBB: c.951T>A , no éxon 8 em P-1 e, c271c>T no éxon 3 em P-4, configurando a DGC ligada ao X. Uma paciente (P-3) apresentou a forma autossômica recessiva, com a mutação c.75_76delGT no éxon 2 do gene NCF1.
Exames radiológicos complementares
Avaliação de sequela pulmonar com tomografia computadorizada (TC) de tórax foi realizada em todos os pacientes ao diagnóstico, e posteriormente a cada dois anos. Todos os pacientes apresentavam alterações no exame, destes, 3/5 desde o diagnóstico: atelectasia (P-1) e pneumonia (P-2 e P-3). Nas imagens radiológicas mais recentes foram observadas: bronquiolite obliterante (P-1 e P-5), bronquiectasias (P-2 e P-3), imagem em vidro fosco (P-1, P-3 e P-4) e nódulos pulmonares (P-1 e P-4).
Seguimento e terapêutica
A profilaxia com SMZ-TMP e antifúngico como preconizado2 foi estabelecida em todos os pacientes. O itraconazol foi o antifúngico de escolha. Cetoconazol e fluconazol foram utilizados inicialmente em dois pacientes (P-1 e P-2) e substituídos posteriormente pelo itraconazol. Anfotericina B lipossomal e voriconazol foram prescritos para tratamentos específicos de fungemia disseminada e pneumonia fúngica em dois pacientes (P-1 e P-2) (Tabela 1). O Interferon-gama foi utilizado em dois pacientes (P-1 e P-4). Todos os pacientes apresentaram algum quadro infeccioso no seguimento, com necessidade de intervenção terapêutica precoce.
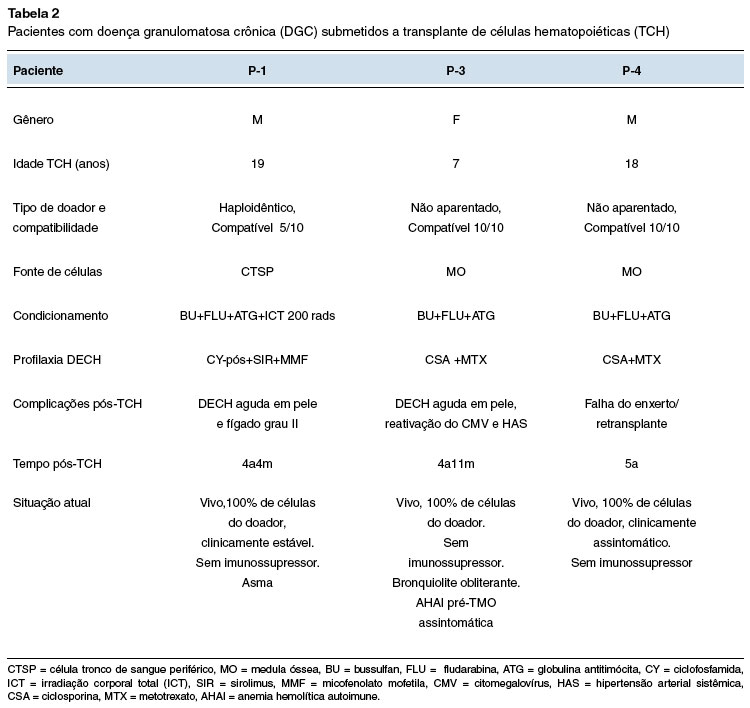
Três pacientes, com defeito genético confirmado e quadro clínico mais grave (P-1, P-3 e P-4) foram submetidos ao TCH no Hospital de Clínicas da Universidade Federal do Paraná, com idades entre 7 a 19 anos (Tabela 2). A sobrevida atual pós-TCH é de: 4 anos e 4 meses (P-1), 4 anos e 11 meses (P-3) e 5 anos (P-4), com pega completa do enxerto e sem medicações imunossupressoras. Um dos pacientes apresentou uma rejeição tardia e foi submetido a um segundo transplante com sucesso. Todos os pacientes têm teste DHR normal pós-TCH.
Discussão
A prevalência da DGC é estimada em 1:200.0002,3, porém em nosso país ainda é desconhecida. Oliveira-Junior e cols. descrevem uma casuística de 71 casos de DGC na América Latina, com 39% de pacientes brasileiros9, um reduzido número de diagnósticos ao se considerar o tamanho da população brasileira e a estatística internacional. No presente estudo foram incluídos cinco pacientes com DGC, a maior parte do sexo masculino e com início dos sintomas no primeiro ano de vida, porém com diagnóstico por volta dos 4 anos de idade. Este dado é relevante, uma vez que a DGC cursa com infecções graves e muitas vezes fatais, ou sequelas que podem comprometer diversos órgãos, em especial o pulmão, em uma fase precoce da vida. Acredita-se que o atraso no diagnóstico possa estar relacionado à dificuldade de acesso dos pacientes a profissionais com conhecimento sobre as manifestações clínicas de EII, tal como observado por Dantas e cols. ao avaliar o conhecimento de médicos sobre os sinais de alerta de imunodeficiência congênita12.
Todos os pacientes apresentaram infecções do trato respiratório, alguns com manifestações graves, como presença de derrame pleural e pneumonia fúngica. A disseminação da infecção fúngica para outros órgãos, principalmente por fungos da espécie Aspergillus sp., tem alta taxa de mortalidade e é frequente nos pacientes com DGC2,10. Neste estudo, entretanto, a espécie C. albicans foi a mais comum, o que pode estar relacionado às características dos pacientes, ou dificuldades técnicas para isolamento de outros fungos. Não houve nenhum caso de abscesso hepático, comumente descrito em pacientes com DGC2,3. Por outro lado, houve isolamento de E. coli e Enterobacter sp., pouco comuns na DGC.
A infecção por micobactérias, seja M. tuberculosis causando a tuberculose (TB) ou M. bovis inoculado pela vacina BGC, fazem parte do espectro clínico da DGC nos países onde a tuberculose é endêmica, como observado neste estudo9,11. A reação adversa grave à BCG é uma manifestação clínica precoce também de outros EII, como a imunodeficiência combinada grave (SCID)9,13, e em países onde a vacina é obrigatória, como o Brasil, tem grande impacto na sobrevida dos pacientes. Desta forma, tem sido proposto postergar a vacina até que defeitos graves do sistema imune possam ser excluídos9,11,13. As alterações pulmonares observadas na TC de tórax geralmente decorrem das diversas pneumonias pré-diagnóstico, bem como de processo inflamatório crônico e podem ter influência na sobrevida e qualidade de vida dos pacientes2,3. Manifestações gastrintestinais observadas em alguns pacientes, tais como granuloma esofágico, são comuns, entretanto não houve casos de colite, divergindo de outros estudos, onde o cólon é frequentemente comprometido2,3,10.
Todos os pacientes receberam profilaxia com SMZ-TMP associado a antifúngico, sem efeitos adversos relevantes. A profilaxia com SMZ-TMP é bem estabelecida há mais de 40 anos, com boa efetividade na redução das infecções2,3. A partir da década de 1990, os antifúngicos, em especial o itraconazol, passaram a fazer parte do arsenal terapêutico da DGC, reduzindo consideravelmente óbitos por pneumonia fúngica2,14.
Três pacientes foram submetidos ao TCH. Estudos sobre TCH para DGC ainda são escassos em nosso país5,6. Há estudos que relatam a indicação de TCH para todos os pacientes com DGC mal controlada e para aqueles com morbidade significativa, como infecções recorrentes com risco de vida e doença pulmonar progressiva14. A sobrevida pós-TCH na DGC tem aumentado devido a vários fatores, como condicionamento com toxicidade reduzida, melhor seleção da fonte de células e doadores, além de avanços na terapia antimicrobiana e menor idade no momento do TCH6,14,15. No entanto, ainda é considerado um procedimento de alto risco associado a morbidade significativa e desfecho fatal, especialmente para pacientes com menos de 14 anos de idade14,15. Entre os pacientes transplantados, dois deles eram adolescentes, com várias comorbidades e alto risco de complicações precoces e de longo prazo. Eles receberam um regime de condicionamento de toxicidade reduzida, com pega do enxerto bem-sucedida. Dois tiveram DECH leve, controlados com uso de corticosteroides e estão sem imunossupressão. O outro paciente desenvolveu uma falha secundária do enxerto que foi resolvida com sucesso após um segundo transplante. Este paciente também está vivo e clinicamente estável após mais de 4 anos do transplante.
Conclusão
A DGC é uma doença grave com início dos sintomas na infância, desta forma, a suspeita diagnóstica e a intervenção terapêutica precoce aumentam a sobrevida dos pacientes. O monitoramento cuidadoso de infecções graves e tratamento imediato são cruciais para a sobrevivência dos pacientes com DGC em longo prazo. Além disso, sugere-se avaliação de alteração pulmonar periódica em todos os casos da doença. Finalmente, O TCH é um tratamento viável para pacientes com DGC no Brasil, mesmo no final da adolescência.
Referências
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019 update on the classification from the International Union of Immunological Societies Expert Committee. Clin Immunol. 2020;40,24-64.
2. Arnold DE, Heimall JR. A Review of Chronic Granulomatous Disease. Adv Ther. 2017;34(12):2543-57.
3. Holland SM. Chronic Granulomatous Disease. Hematol Oncol Clin North Am. 2013 Feb;27(1):89-99, viii.
4. ESID Registry - Working definitions for clinical diagnosis of PID [internet]. Disponível em: https://esid.org/Working-Parties/Registry-Working-Party/Diagnosis-criteria.
5. Fernandes JF, Kerbauy FR, Ribeiro AAF, Kutner JM, Camargo LFA, A Stape, et al. Transplante alogênico de células-tronco hematopoéticas em crianças com imunodeficiências primárias: a experiência do Hospital Israelita Albert Einstein. Einstein. 2011 Apr 1;9(2 Pt 1):140-4.
6. Fernandes JF, Nichele S, Arcuri LJ, Ribeiro L, Zamperlini-Netto G, Loth G, et al. Outcomes after Haploidentical Stem Cell Transplantation with Post-Transplantation Cyclophosphamide in Patients with Primary Immunodeficiency Diseases. Biol Blood Marrow Transplant. 2020 Oct;26(10):1923-9.
7. Fujimura MD. Níveis séricos das subclasses de IgG em crianças normais e nefróticas [tese]. São Paulo: Faculdade de Medicina, Universidade de São Paulo; 1991.
8. Moraes-Pinto MI, Ono E, Santos-Valente EC, Almeida LC, Andrade PR, Dinelli MI, et al. Lymphocyte subsets in human immunodeficiency virus-unexposed Brazilian individuals from birth to adulthood. Mem Inst Oswaldo Cruz. 2014;109(8):989-98.
9. Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke L, et al. Clinical and Genotypic Spectrum of Chronic Granulomatous Disease in 71 Latin American Patients: First Report from the LASID Registry. Pediatr Blood Cancer. 2015 Dec;62(12):2101-7.
10. van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L, et al. Chronic Granulomatous Disease: the European experience. PLoS One. 2009;4(4):e5234.
11. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira Jr E, et al. Mycobacterial disease in patients with Chronic Granulomatous Disease: a retrospective analysis of 71 cases. J Allergy Clin Immunol. 2016 Jul;138(1):241-248.e3.
12. Dantas EO, Aranda CS, Nobre FA, Fahl K, Mazzucchelli JTL, Felix E, Friedlander-Del Nero DL, et al. Conhecimento médico sobre as imunodeficiências primárias na cidade de São Paulo, Brasil. Einstein. 2013;11(4):479-85.
13. Mazzucchelli JTL, Bonfim C, Castro G, Condino-Neto A, Costa NMX, Cunha L. Severe Combined Immunodeficiency in Brazil: management, prognosis, and BCG-Associated complications. J Investig Allergol Clin Immunol. 2014;24(3):184-91.
14. Segal BH, Veys P, Malech H, Cowan MJ. Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant. 2011 Jan;17(1 Suppl):S123-31.
15. Connelly JA, Marsh R, Parikh S, Talano JA. Allogeneic hematopoietic cell transplantation for Chronic Granulomatous Disease: controversies and state of the art. J Pediatric Infect Dis Soc. 2018 May 9;7 (Suppl 1):S31-S39.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888