Número Atual: Outubro-Dezembro 2021 - Volume 5 - Número 4
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicações Clínicas e Experimentais
Apresentação atípica de síndrome de hiper-IgM ligada ao X simulando doença inflamatória intestinal
Atypical presentation of X-linked hyper-IgM syndrome simulating inflammatory bowel disease
Nara Lillian Lima Cardoso1; François Loiola Ponte de Souza1 2; Hildenia Baltasar Ribeiro Nogueira3; Janáira Fernandes Severo Ferreira4; Tábata Takahashi França5; Antonio Condino-Neto5
DOI: 10.5935/2526-5393.20210063
1. Hospital Infantil Albert Sabin, Residência médica em Pediatria - Fortaleza, CE, Brasil
2. Hospital Infantil Albert Sabin, Supervisor da Residência Médica em Pediatria - Fortaleza, CE, Brasil
3. Hospital Infantil Albert Sabin, Serviço de Gastroenterologia Pediátrica - Fortaleza, CE, Brasil. Universidade de Fortaleza - Fortaleza, CE, Brasil
4. Hospital Infantil Albert Sabin, Serviço de Imunologia Pediátrica - Fortaleza, CE, Brasil
5. Universidade de São Paulo, Instituto de Ciências Biomédicas - São Paulo, SP, Brasil
Endereço para correspondência:
Nara Lillian Lima Cardoso
E-mail: nara_lillian@hotmail.com / janairafs@gmail.com
Submetido em: 06/07/2021
Aceito em: 12/10/2021
Não foram declarados conflitos de interesse associados à publicação deste artigo
RESUMO
Relatamos o caso de um paciente do sexo masculino, que iniciou quadro de úlceras em trato gastrointestinal, associado a febre recorrente e diarreia com muco e sangue aos 10 meses de vida, suspeitado inicialmente de doença inflamatória intestinal, no entanto, não apresentou melhora do quadro com terapia imunossupressora, sendo realizada investigação para erro inato da imunidade. Nos exames laboratoriais, apresentou níveis baixos de IgG e IgA e níveis elevados de IgM e neutropenia persistente. Diante disso, foi realizado teste genético que confirmou diagnóstico de síndrome de hiper-IgM ligada ao X. Os erros inatos da imunidade podem se manifestar com doenças do trato gastrointestinal, de forma relativamente frequente, devendo entrar como diagnóstico diferencial de diarreia crônica. Inclusa nesse grupo de doenças, as síndromes de hiper-IgM constituem um grupo heterogêneo de doenças, possuindo em comum níveis significativamente baixos ou ausentes de IgG e IgA e níveis normais ou elevados de IgM, o que predispõe a infecções e febre recorrente; além de outras alterações laboratoriais, como neutropenia, que pode estar associada a úlceras no trato gastrointestinal e proctite, simulando apresentação clínica de doença inflamatória intestinal. Para o paciente relatado, foi iniciada terapia com imunoglobulinas de forma periódica, além de antibioticoprofilaxia para infecções, evoluindo com resposta clínica satisfatória. O artigo possui objetivo principal de alertar para o diagnóstico diferencial de erros inatos da imunidade diante do quadro apresentado, visando o diagnóstico precoce e a instituição da terapia adequada.
Descritores: Doenças da imunodeficiência primária, doenças do sistema imunitário, síndrome de imunodeficiência com hiper-IgM tipo 1.
INTRODUÇÃO
Os erros inatos da imunidade (EII) são distúrbios genéticos que afetam diferentes componentes do sistema imune. Atualmente, devido ao aprimoramento dos métodos de diagnóstico genético, são descritas mais de 400 doenças. No entanto, ainda é comum o diagnóstico tardio ou de forma incorreta1,2. As manifestações clínicas são muito diversas, caracterizando-se por infecções graves recorrentes ou prolongadas, doença autoimune/inflamatória, alergia ou malignidade3,4.
Os EII podem acometer o trato gastrointestinal em uma frequência que varia de 5 a 50%1. O tecido linfoide associado ao intestino é o maior órgão linfoide do corpo, com mecanismos variados de regulação imunológica. Diarreia e má absorção são comuns em muitos EII. Doenças gastrointestinais recorrentes ou refratárias ao tratamento devem ser um sinal de alerta para uma possível imunodeficiência5.
A síndrome de hiper-IgM (HIGM) pode ser congênita ou secundária a outras doenças de base (mieloma múltiplo, leucemia, síndrome nefrótica, e infecções crônicas, como síndrome da rubéola congênita e uso de medicamentos como fenitoína)6. As formas congênitas são muito raras, correspondendo de 0,3 a 2,9% de todas as imunodeficiências primárias, tendo uma incidência estimada em 1/130.000 nascidos vivos e com uma heterogeneidade de defeitos genéticos, podendo apresentar herança ligada ao X, autossômica recessiva ou dominante3,7,8. Na HIGM ocorre defeito na troca de classes das imunoglobulinas devido a defeitos genéticos na via de sinalização do CD40 (linfócito B)/ CD40 ligante (CD40L; linfócitos T) ou no sistema de reparo do DNA responsável pela troca de classe. Portanto, há um prejuízo na sinalização necessária para que linfócitos T ativados induzam os linfócitos B a converter imunoglobulina M (IgM) em outras imunoglobulinas (IgG, IgA e IgE)2,9,10. Além disso, o CD40L também participa na maturação de células apresentadoras de antígenos, na estimulação da função efetiva de macrófagos e no aprimoramento dos antígenos de linfócitos T8,11,12.
A depender do defeito genético associado, a HIGM pode ser classificada em cinco subtipos: tipo 1, ocorre devido à deficiência de CD40L, é hereditária autossômica dominante ligada ao X, sendo exclusiva do sexo masculino e a forma mais comum, correspondendo a 65% dos casos5; tipo 2, corresponde à forma autossômica recessiva com mutações no gene que codifica uma citidina desaminase que participa da cascata de ativação intracelular do linfócito B5. Estes pacientes podem ter hiperplasia adenoide com defeito nos centros germinativos, representando cerca de 15% dos casos; tipo 3, a mutação ocorre no gene que codifica especificamente para a molécula de CD40 essencial no desenvolvimento, crescimento e diferenciação dos linfócitos; tipo 4, cujos mecanismos moleculares ainda são desconhecidos; e tipo 5, produzido por mutações no gene para uma glicosilase (uracil DNA glicosilase), sendo os dois últimos formas recessivas6. Todas essas síndromes apresentam características clínicas semelhantes e somente por meio de estudos moleculares e genéticos torna-se possível um diagnóstico diferencial1,13. Os pacientes apresentam como características níveis significativamente baixos ou ausentes de IgG e IgA e níveis normais ou elevados de IgM, além de uma resposta de IgG às vacinações fraca ou não protetora14. Neutropenia é a alteração hematológica mais comum na HIGM do tipo 1, porém sua causa permanece desconhecida, podendo ser devido à presença de anticorpos antineutrófilos e/ou por atraso da maturação mieloide na medula15. Alguns estudos sugerem que o CD40-ligante possa atuar também na estimulação da produção endógena de estimulador de colônia de granulócitos15, e a biópsia da medula óssea desses pacientes pode mostrar atraso na maturação da linhagem mieloide14.
A maioria dos pacientes com HIGM apresenta aumento de susceptibilidade a infecções, principalmente sinopulmonares, como pneumonia, sinusite e otite média aguda, desenvolvendo sintomas durante o primeiro ano de vida; e quase todos durante os primeiros quatro anos16. A pneumonia é a infecção mais prevalente e em metade dos casos é causada por Pneumocystis jirovecii16. Complicações infecciosas do trato respiratório, como bronquiectasias, são comuns9. A diarreia infecciosa tem sido associada à infecção por cryptosporidium, giardia, salmonella ou entamoeba2. Úlceras aftosas, gengivite e úlceras retais podem estar associadas à neutropenia crônica ou intermitente2. Também podem acontecer infecção do sistema nervoso central, sepse, hepatite e/ou colangite esclerosante, celulite e/ou abscessos subcutâneos16. Devido a infecções recorrentes, esses pacientes podem apresentar falha do crescimento e desenvolvimento16,17. Há risco aumentado de neoplasias, principalmente de fígado e de vias biliares, e de complicações autoimunes, como colangite esclerosante, que podem estar associados à infecção crônica por Cryptosporidium parvum 9,16,18.
Relatamos o caso de um paciente do sexo masculino que apresentou sinais de alarme para erro inato da imunidade, com infecção grave, além de febre recorrente, diarreia crônica, úlceras orais e neutropenia, sendo inicialmente manejado como doença inflamatória intestinal, no entanto, com o diagnóstico posterior confirmado de HIGM tipo 1, com o objetivo de fazer lembrar dessa hipótese diagnóstica diante de quadros semelhantes.
RELATO DE CASO
Paciente do sexo masculino, nascido de parto cesáreo, a termo, pais não consanguíneos, uma irmã saudável, sem intercorrências no período perinatal, aleitamento misto desde do nascimento e introdução alimentar aos 6 meses, apresentando crescimento e desenvolvimento normais. Primeiro internamento aos 6 meses de idade devido pneumonia grave, evoluindo com hipossaturação e desconforto respiratório importante, com necessidade de internamento em unidade de terapia intensiva, com realização de intubação orotraqueal e boa resposta a antibioticoterapia de amplo espectro (piperacilina-tazobactam e vancomicina). Ficou assintomático até 10 meses de vida, quando iniciou quadro de úlceras em cavidade oral associado à febre diária, principalmente noturno, procurando atendimento médico algumas vezes, sendo realizada antibioticoterapia com amoxicilina e amoxicilina-clavulanato, sendo relatado de acordo com a genitora melhora do quadro com antibioticoterapia, no entanto, retorno logo após o término da medicação. Durante esse período, chegou a apresentar quadro descrito como abscesso dentário, como complicação das úlceras, com melhora após uso de antibiótico. Aos 12 meses de vida, iniciou com episódios de diarreia com sangue e com muco, 3-4 vezes ao dia e placas avermelhadas evoluindo para manchas hipercrômicas mais intensas em membros inferiores e em joelhos, e foi interrogado artralgia (dificuldade de apoiar pés no chão). Foi tratado com antibiótico e corticoide, com boa resposta.
Por duas vezes foi internado devido ao quadro de úlceras orais, febre e diarreia com sangue, sendo tratado com antibioticoterapia. Foi submetido à dieta de exclusão da proteína do leite de vaca, entretanto não apresentou melhora, e após reintrodução dessa proteína, não houve alterações do quadro intestinal.
Dessa forma, o serviço de gastroenterologia levantou hipótese de doença inflamatória intestinal, sendo realizada endoscopia digestiva alta que mostrou úlcera esofágica rasa, sem alterações microscópicas significativas e colonoscopia que evidenciou úlceras isoladas, rasas, com mucosa adjacente enantemática e edemaciadas em colo transverso, colo esquerdo, sigmoide e reto, sem atividade inflamatória a microscopia. Foi iniciada terapia para doença inflamatória intestinal com prednisona, azatioprina, sulfassalazina e fórmula enteral adequada. No entanto, persistia com febre de padrão intermitente, úlceras orais e perianais e diarreia com sangue e muco, com necessidade de internamentos no período para realização de antibioticoterapia endovenosa, com melhora do quadro. Aos 15 meses, internado por quadro de diarreia tipo colite, suspeita de EII e foi solicitado avaliação do serviço de Imunologia, tendo sido a hipótese inicial uma deficiência de IL-10/receptor de IL-10 e solicitado painel genético para EII. Paralelamente, foram solicitados outros exames imunológicos e dosagem de imunoglobulinas, evidenciando IgG e IgA abaixo do percentil 3 e IgM acima do percentil 97. A dosagem de imunoglobulinas foi repetida, mantendo IgA e IgG abaixo percentil 3 (P3) para idade e IgM normal, sendo iniciado reposição de imunoglobulina endovenosa, suspenso corticoide e imunossupressor e mantida terapia antimicrobiana com melhora clínica e laboratorial. Na revisão de hemogramas, observou-se anemia intermitente e neutropenia persistente desde 12 meses de vida, sendo realizado também mielograma com biópsia óssea, mostrando hipocelularidade eritroide e granulocítica com moderado retardo da maturação mieloide. O paciente recebeu terapia com estimulador de colônias de granulócitos, com melhora importante da neutropenia. A dosagem de vitamina B 12 e de ácido fólico estavam normais e outros exames já tinham sido realizados como sorologias para Epstein-barr vírus e citomegalovírus com IgG e IgM negativos, anti-HIV negativo, além de pANCA e cANCA, calprotectina fecal, anti-tripsina fecal, HLAB51 negativos. Durante os internamentos, foi isolado Campylobacter jejunii em PCR multiplex das fezes, Klebsiella e Citrobacter freundii em urinoculturas.
Aos 19 meses de idade, o resultado do painel genético para EII (407 genes investigados - laboratório Invitae) indicou mutação no CD40 ligante (Figura 1). Posteriormente, foi realizada citometria de fluxo que mostrou alteração na expressão da proteína do CD40L (Figura 2), e a amplificação do produto de PCR em gel de agarose mostrou ausência de amplificação dos exons 4 e 5 (Figura 3), confirmando o diagnóstico de HIGM ligada ao X.

Figura 1
Painel genético para erros inatos da imunidade (EII - 407 genes)
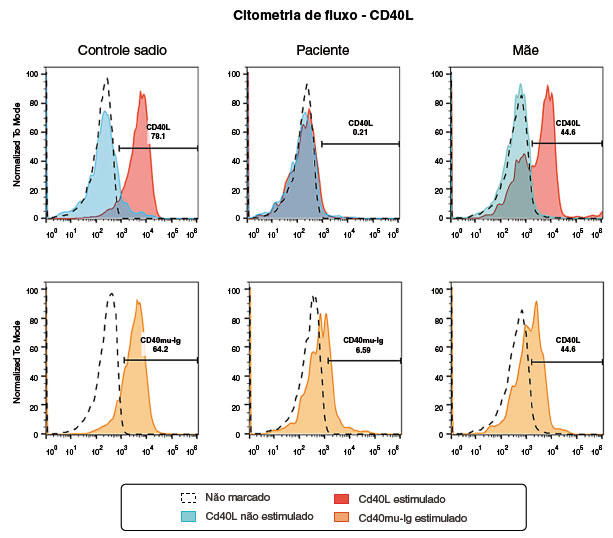
Figura 2
Expressão proteína CD40 Ligante por citometria de fluxo
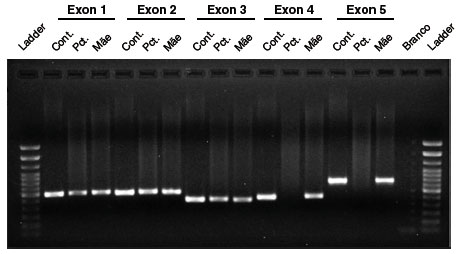
Figura 3
Amplificação dos exons do CD40LG em gel de agarose
Não foram observadas mutações patogênicas nos demais genes.
DISCUSSÃO
A HIGM ligada ao X ou tipo 1 caracteriza-se por deficiência do CD40L, que acomete indivíduos do sexo masculino, em geral, filhos de mães portadoras da mutação em um dos cromossomos X19. Estes pacientes apresentam um defeito da função de células B e de células T, sendo esta considerada uma imunodeficiência primária combinada20. O paciente do caso relatado é do sexo masculino e apresentou sinais de imunodeficiência primária durante o primeiro ano de vida, devido pneumonia grave, com necessidade de intubação orotraqueal. As infecções sinopulmonares, principalmente a pneumonia, estão presentes em 80% dos pacientes com HIGM ligada ao X, ocorrendo durante o primeiro ano de vida16. Aproximadamente metade dos pacientes com HIGM apresentam pneumonias causadas por P. jirovecii16. Por ser uma imunodeficiência combinada, estes pacientes além de apresentarem susceptibilidade a infecções por bactérias oportunistas, como P. jirovecii e histoplasmose, também apresentam maior predisposição a infecções por bactérias encapsuladas, como Streptococcus pneumonie ou Haemophilus influenzae, prováveis causadores da pneumonia apresentada pelo paciente, além de apresentar quadro de febre recorrente com melhora após uso de antibioticoterapia e PCR multiplex das fezes isolando bactéria, confirmando a maior predisposição a infecções16,17.
O paciente apresentou, ainda, diarreia crônica, sendo uma manifestação comum da síndrome de Hiper-IgM ligada ao X e mais comumente decorrente de infecções por cryptosporidium16. A diarreia associada a úlceras do trato gastrointestinal poderia ser justificada também pela neutropenia persistente, considerada um achado hematológico comum presente nessa síndrome, presente em dois terços a metade dos pacientes, podendo ser episódica ou recorrente, estando associada a úlceras no trato gastrointestinal, estomatite e proctite, além de aumentar o risco de infecções12,14,16. A biópsia da medula óssea desses pacientes pode mostrar atraso na maturação da linhagem mieloide, como no caso relatado14.
Inicialmente foi suspeitada doença inflamatória intestinal de início precoce; no entanto, o paciente não apresentava marcadores inflamatórios compatíveis com diarreia inflamatória, como calprotectina e alfa-1-antitripsina fecal, além de não apresentar microscopia sugestiva nas biópsias das úlceras e não apresentar boa resposta com o uso de imunossupressores. Além disso, os exames laboratoriais evidenciaram IgM em níveis normais a altos e IgE, IgG e IgA em níveis baixos, além de neutropenia. Diante disso, foi suspeitada imunodeficiência primária, sendo confirmado o diagnóstico de HIGM ligada ao X através do teste genético.
As opções terapêuticas utilizadas englobam reposição de imunoglobulina endovenosa, antibioticoprofilaxia para infecção pelo P. jirovecii com sulfametoxazol-trimetropim, uso de estimulador de colônia de granulócitos para neutropenia e transplante de medula óssea, com graus variados de sucesso3,4,15,16,20. Além disso, não é recomendado que esses pacientes recebam vacinas de vírus vivos, e deve ser recomendada prevenção de infecção por cryptosporidium (contaminação da água), com medidas higiênicas, como beber apenas água filtrada, não ter contato com fezes e evitar banhos em lagos, lagoas e em piscinas não cloretadas7,12,14. A única terapêutica curativa é o transplante alogênico de células hematopoiéticas, com melhores resultados em pacientes jovens, sem doença hepática no momento do transplante, e com realização de boa supressão medular, devendo ser uma opção terapêutica considerada9,14. No caso do paciente relatado, foi observada melhora clínica e laboratorial com uso de imunoglobulina IgG endovenosa de forma regular, além de bom controle da diarreia, das úlceras em trato gastrointestinal e da neutropenia com uso de estimulador de colônia de granulócitos e de sulfametoxazol trimetropim profilático; foram orientadas também medidas higiênicas para a genitora.
CONCLUSÃO
A HIGM ligada ao X deve ser lembrada quando há aumento de susceptibilidade a infecções, podendo se manifestar na forma de febre recorrente, diarreia crônica e/ou múltiplos internamentos por quadro infecciosos, associado à redução de imunoglobulinas IgG, IgA e IgE, com imunoglobulina IgM normal ou aumentada e neutropenia, principalmente em pacientes do sexo masculino. A principal causa de morte desses pacientes são infecções oportunistas, a partir daí, a importância do diagnóstico precoce e da instituição das profilaxias adequadas, além de mais precocemente poder programar terapia curativa com transplante alogênico de células hematopoiéticas.
AGRADECIMENTOS
A Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) apoiou T.T.F. (processo n° 2017/04187-9) e A.C.N. (processo n° 2016/22158-3). The Hyper IgM Foundation apoiou T.T.F. e A.C.N., The Jeffrey Modell Foundation apoiou A.C.N.
REFERÊNCIAS
1. Amaya-Uribe L, Rojas M, Azizi G, Anaya JM, Gershwin ME. Primary immunodeficiency and autoimmunity: A comprehensive review. J Autoimmun. 2019 May;99:52-72.
2. Wu J, Zhong W,YinY, Zhang H. Primary immunodeficiency disease: a retrospective study of 112 Chinese children in a single tertiary care center. BMC Pediatr. 2019 Nov 4;19(1):410.
3. Justiz Vaillant AA, Qurie A. Immunodeficiency. 2021 Jun 30. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan-. PMID: 29763203.
4. Benkerrou M, Gougeon ML, Griscelli C, Fischer A. Hypogammaglobulinémie G et A avec hypergammaglobulinémie M. A propos de 12 observations [Hypogammaglobulinemia G and A with hypergammaglobulinemia M. Apropos of 12 cases]. Arch Fr Pediatr. 1990 May;47(5):345-9. French. PMID: 2369267.
5. Agarwal S, Cunningham-Rundles C. Gastrointestinal Manifestations and Complications of Primary Immunodeficiency Disorders. Immunol Allergy Clin North Am. 2019 Feb;39(1):81-94. doi: 10.1016/j.iac.2018.08.006.
6. de la Morena MT. Clinical Phenotypes of Hyper-IgM Syndromes. Clinical Management Review. 2016;1023-34.
7. de la Morena MT. Clinical Phenotypes of Hyper-IgM Syndromes. J Allergy Clin Immunol Pract. 2016 Nov-Dec;4(6):1023-1036. doi: 10.1016/j.jaip.2016.09.013.
8. Saiki O, Tanaka T, Wada Y, Uda H, Inoue A, Katada Y, et al. Signaling through CD40 rescues IgE but not IgG or IgA secretion in X-linked immunodeficiency with hyper-IgM. J Clin Invest. 1995 Feb;95(2):510-4. doi: 10.1172/JCI117692.
9. Yazdani R, Fekrvand S, Shahkarami S, Azizi G, Moazzami B, Abolhassani H, et al. The hyper IgM syndromes: Epidemiology, pathogenesis,clinicalmanifestations,diagnosisandmanagement.Clin Immunol. 2019 Jan;198:19-30. doi: 10.1016/j.clim.2018.11.007.
10. Kim D, Shin JA, Han SB, Chung NG, Jeong DC. Pneumocystisjirovecii pneumonia as an initial manifestation of hyper-IgM syndrome in an infant: A case report. Medicine (Baltimore). 2019 Feb;98(7):e14559. doi: 10.1097/MD.0000000000014559.
11. Notarangelo LD, Duse M, Ugazio AG. Immunodeficiency with hyper-IgM (HIM). Immunodefic Rev. 1992;3(2):101-21. PMID: 1554497.
12. Ameratunga R, Woon ST, Koopmans W, French J. Cellular and molecular characterisation of the hyperimmunoglobulin M syndrome associated with congenital rubella infection. J Clin Immunol. 2009;29:99.
13. Groeneweg M, Hartwig NG, Poerink-Stockschlãder AB, Schweizer JJ, Bijleveld CM, Bredius RG. Twee kinderen met ernstige, recidiverende infecties en het X-gebonden hyper-IgM-syndroom [Two children with severe recurrent infections and the X-linked hyper-IgM syndrome]. Ned Tijdschr Geneeskd. 2003 May 24;147(21):1024-8. Dutch. PMID: 12811975.
14. Yazdani R, Fekrvand S, Shahkarami S, Azizi G, Moazzami B, Abolhassani H, Aghamohammadi A. The hyper IgM syndromes: Epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol. 2019 Jan;198:19-30. doi: 10.1016/j. clim.2018.11.007.
15. Atarod L, Aghamohammadi A, Moin M, Kanegane H, Rezaei N, Rezaei Kalantari K, et al. Successful management of neutropenia in a patient with CD40ligand deficiency byimmunoglobulin replacement therapy. Iran J Allergy Asthma Immunol. 2007 Mar;6(1):37-40. PMID: 17303928.
16. Winkelstein JA, Marino MC, Ochs H, Fuleihan R, Scholl PR, Geha R, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Baltimore). 2003 Nov;82(6):373-84. doi: 10.1097/01.md.0000100046.06009.b0.
17. Levy J, Espanol-Boren T, Thomas C, Fischer A,Tovo P, Bordigoni P, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997 Jul;131(1 Pt 1):47-54. doi: 10.1016/s0022-3476(97)70123-9.
18. Van Hoeyveld E, Zhang PX, De Boeck K, Fuleihan R, Bossuyt X. Hyper-immunoglobulin M syndrome caused by a mutation in the promotor for CD40L. Immunology. 2007 Apr;120(4):497-501. doi: 10.1111/j.1365-2567.2006.02520.x. Epub 2007 Jan 17. PMID: 17244160.
19. Lougaris V, Badolato R, Ferrari S, Plebani A. Hyper immunoglobulin M syndrome due to CD40 deficiency: clinical, molecular, and immunological features. Immunol Rev. 2005 Feb;203:48-66. doi: 10.1111/j.0105-2896.2005.00229.x. PMID: 15661021.
20. Wang WC, Cordoba J, Infante AJ, Conley ME. Successful treatment of neutropenia in the hyper-immunoglobulin M syndrome with granulocyte colony-stimulating factor. Am J Pediatr Hematol Oncol. 1994 May;16(2):160-3. PMID: 7513136.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888