Número Atual: Abril-Junho 2021 - Volume 5 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Imagens em Alergia e Imunologia
Angioedema hereditário inaugural na idade pediátrica
Hereditary angioedema in a child with no family history of disease
Marisa Paulino; Célia Costa
DOI: 10.5935/2526-5393.20210034
Hospital de Santa Maria - Centro Hospitalar Universitário Lisboa Norte, Serviço de Imunoalergologia - Lisboa, Lisboa, Portugal
Endereço para correspondência:
Marisa Paulino
E-mail: marisapaulino.90@gmail.com
Submetido em: 01/02/2021
Aceito em: 24/03/2021
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
O angioedema hereditário por défice de C1-inibidor é uma doença rara autossômica dominante com uma prevalência estimada em 1:50.000. Habitualmente a história familiar aponta para este diagnóstico. No entanto, a apresentação atípica com história familiar negativa pode atrasar o diagnóstico de meses a anos. Os autores apresentam o caso de uma criança de 6 anos sem antecedentes pessoais ou familiares relevantes que recorreu ao Serviço de Urgência pediátrico por edema, calor e rubor do cotovelo, joelho e maléolos direitos com 12h de evolução, sem fatores associados. Ao exame objetivo: edema do cotovelo, joelho e maléolos direitos, exantema não pruriginoso maleolar homolateral com discreto desconforto à palpação. Sem elevação dos parâmetros infeciosos ou inflamatórios. Foi iniciada corticoterapia sistêmica, com melhoria lenta do quadro. Teve alta, referenciada à consulta de Imunoalergologia. Na anamnese foram apurados quatro episódios de edema periarticular nos doze meses prévios. A avaliação analítica da criança revelou C1 inibidor 62 mg/dL, C1 inibidor funcional 29%, confirmada em duas determinações, e a dos pais e dos dois irmãos foi normal. No estudo genético não foram identificadas mutações nos genes SERPING. O angioedema hereditário por défice de função do C1-inibidor -tipo II -representa 15 a 20% dos casos. Embora a história familiar seja o maior sinal de alerta para o diagnóstico desta patologia, em 20-25% dos casos ocorre mutação espontânea. Nestes casos um elevado grau de suspeição é necessário e um atraso no diagnóstico pode levar a consequências graves. As opções terapêuticas em crianças menores de 12 anos são ainda limitadas.
Descritores: Angioedema, angioedema hereditário tipos I e II, diagnóstico, pediatria.
INTRODUÇÃO
O angioedema hereditário por défice do inibidor da protease C1 plasmática (C1-INH) é uma doença autossómica dominante rara que afeta uma em cada 50.000 pessoas1,2. É causado por mutações no gene SERPING 1 que codifica o C1-INH. Estão descritas mais de 400 mutações do gene SERPING vasoativas, como a bradicinina, aumentando a permeabilidade vascular levando à formação de edema1,2. Habitualmente manifesta-se na infância, com a média de idade de início estimada aos 12 anos. Cerca de 90% dos doentes tem o seu primeiro episódio antes dos 20 anos4-6.
O angioedema hereditário é caracterizado por episódios recorrentes de angioedema subcutâneo ou submucoso (sem urticária associada), da face, via aérea superior, sistema gastrointestinal, pele e vias geniturinárias7. Frequentemente associam-se pródromos, dos quais o eritema marginatum - um exantema serpiginoso não pruriginoso, no local do edema - ocorre em 1/3 dos casos3. Astenia, adinamia, e parestesias no local do edema podem preceder as crises3.
Embora a história familiar seja um dos principais fatores que levam à suspeita desta patologia, esta pode não estar presente8. O diagnóstico é feito com base no doseamento da concentração sérica de C1INH e percentagem de C1-INH funcional. Os níveis de C4 podem também estar reduzidos1. As alterações devem ser confirmadas com uma segunda avaliação laboratorial1,8.
DESCRIÇÃO DE CASO
Descrevemos o caso de uma criança de sexo feminino com 6 anos de idade que recorreu ao Serviço de Urgência Pediátrico (SUP) por edema, calor e rubor do cotovelo, joelho e maléolos direitos. Estes sintomas apresentavam 12 horas de evolução e não responderam a tratamento com anti-inflamatório nãoesteroide (AINE) oral (Ibuprofeno). Ao exame objetivo apresentava edema do cotovelo, joelho e maléolos direitos, exantema não pruriginoso maleolar homolateral (Figura 1) e com discreto desconforto à palpação. Não foi encontrado qualquer fator desencadeante. Da avaliação laboratorial no SUP não se verificaram alterações, incluindo parâmetros inflamatórios normais. Após administração de anti-histamínico H1 de segunda geração oral, não se verificou melhoria. Foi admitida no serviço de observação do SUP e iniciada corticoterapia sistêmica com prednisolona 1,5 mg/ kg/dia que manteve durante 3 dias, com melhoria progressiva do quadro. Ficou orientada para consulta de Imunoalergologia.
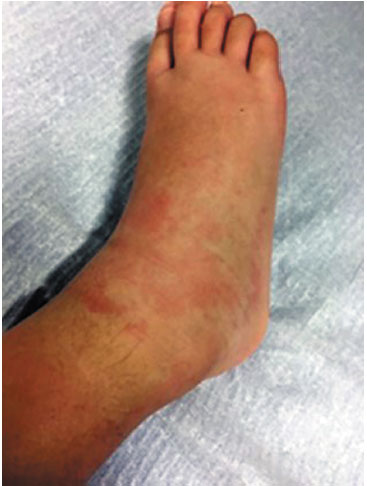
Figura 1
Edema do pé + Eritema marginatum
Na consulta de Imunoalergologia, na anamnese apuraram-se quatro episódios anteriores de edema subcutâneo, com início nos quatro meses anteriores, dois destes acompanhados de dor abdominal e que se seguiram a extrações dentárias. Estes episódios tiveram a duração de 2-3 dias e aparentemente melhoraram após toma de AINE oral (Ibuprofeno). Sem história familiar de angioedema recorrente.
Foi feita avaliação analítica em que se detetou: C1 inibidor 62 mg/dL (21-38), C1 inibidor funcional 29% (> 68%), C4 3,1 mg/dL (15-20). Estes resultados foram confirmados numa segunda determinação, corroborando o diagnóstico de angioedema hereditário tipo 2. Os familiares próximos (mãe, pai, e dois irmãos) foram rastreados, não tendo sido registadas alterações analíticas. Foi efetuado estudo genético com pesquisa de mutações do gene C1 INH (SERPING 1), por sequenciação direta dos exões 1, 2, 3, 4, 5, 6, 7 e 8, e do gene F12, por sequenciação direta do exão 9, não tendo sido detectadas mutações na doente ou nos seus familiares diretos.
Por manter episódios de edema recorrentes (1-2 por mês) associados a situações de stress emocional ou traumatismos minor, com necessidade de recorrer ao SUP para administração de concentrado de C1-INH, foi medicada com profilaxia em longo prazo com ácido tranexâmico 500 mg/dia. Desde esta altura (cerca de 1 ano), sem novos episódios de angioedema.
DISCUSSÃO
O angioedema hereditário tipo II - representa 15 a 20% dos casos de angioedema hereditário1,3. Em 20-25% dos casos a mutação ocorre de forma espontânea, e nestes casos a história familiar está ausente3. Embora não seja essencial para o diagnóstico, a caracterização da mutação genética em doentes com doenças raras permite compreender o potencial patogênico destas mutações7,8.
O eritema marginatum é mais frequente na população pediátrica e é confundido com urticária, podendo levar a erros e atraso no diagnóstico. Sobretudo no sexo feminino, a puberdade pode levar a um aumento da gravidade e frequência das crises.
Frequentemente, a subvalorização dos sintomas ou diagnóstico incorreto levam a atrasos em anos na identificação e tratamento destes doentes. Um estudo de Zanichelli e cols.9 demonstra que cerca de 42% com história familiar positiva são mal diagnosticadas nas primeiras manifestações e esse número é maior, ainda, quando a história familiar está ausente, chegando aos 65%. Um doente não diagnosticado ou incorretamente diagnosticado não só corre riscos de administração de terapêuticas e cirurgias desnecessárias como tem cerca de 30% maior risco de morte por asfixia causada por crises laríngeas6.
A terapêutica em crianças é limitada em relação a adultos. Em situações agudas, o concentrado de C1-INH é a terapêutica de escolha para crianças de qualquer idade1,6-8. Recentemente, como alternativa o icatibant, um antagonista dos recetores ß2 da bradicinina, foi aprovado para crianças maiores de 2 anos6. É, no entanto, na terapêutica de manutenção que a idade pediátrica tem maior condicionamento. Androgênios atenuados, como danazol e estanozolol, estão contraindicados em crianças antes do estadio 5 de Tanner1,8. O concentrado de C1-INH administrado por via subcutânea é a terapêutica preventiva de escolha para crianças, mas não está disponível em Portugal. Sempre que não esteja disponível o ácido tranexâmico deve ser a alternativa1,6-8.
Apesar de ser cada vez maior, é importante continuar a divulgação desta patologia na comunidade médica, sobretudo entre especialidades com componente pediátrico. Em casos em que a suspeita é elevada pela história clínica sugestiva, mesmo na ausência de história familiar, devem ser avaliados os níveis séricos de C1-INH quantitativo e funcional. Casos como o apresentado, em que não são reconhecidas as mutações, podem ser devido a mutações ainda não identificadas pelos testes atualmente praticados, e mostram a importância de existir uma centralização de doentes de modo a otimizar o conhecimento desta patologia. Através da identificação de biomarcadores genéticos que se correlacionem com a gravidade da doença podemos melhorar os cuidados e prevenir complicações nestes doentes10.
REFERÊNCIAS
1. Maurer M, Magerl M, Ansotegui I, Aygören-Pürsün E, Betschel S, Bork K, et al. The international WAO/EAACI guideline for the management of hereditary angioedema - The 2017 revision and update. Allergy Eur J Allergy Clin Immunol. 2018;73(8):1575-96.
2. Savarese L, Bova M, De Falco R, Guarino MD, De Luca Picione R, Petraroli A, et al. Emotional processes and stress in children affected by hereditary angioedema with C1-inhibitor deficiency: A multicenter, prospective study. Orphanet J Rare Dis. 2018;13:115-23.
3. Craig T. Hereditary angioedema. Pediatr Allergy, Immunol Pulmonol. 2014;27(4):157-8.
4. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: New findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-74.
5. Roche O, Blanch A, Caballero T, Sastre N, Callejo D, López-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: Patient registry and approach to the prevalence in Spain. Ann Allergy, Asthma Immunol. 2005;94(4):498-503.
6. Johnston DT, Smith RC. Hereditar y angioedema: Special considerations in children. Allergy Asthma Proc. 2020;41(Suppl 1):S43-6.
7. Pancholy N, Craig T. Hereditary angioedema in children: A review and update. Curr Opin Pediatr. 2019;31(6):863-8.
8. Farkas H, Martinez-Saguer I, Bork K, Bowen T, Craig T, Frank M, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy Eur J Allergy Clin Immunol. 2017;72(2):300-13.
9. Zanichelli A, Longhurst HJ, Maurer M, Bouillet L, Aberer W, Fabien V, et al. Misdiagnosis trends in patients with hereditary angioedema from the real-world clinical setting. Ann Allergy, Asthma Immunol. 2016;117(4):394-8.
10. Germenis AE, Speletas M. Genetics of Hereditary Angioedema Revisited. Clin Rev Allergy Immunol. 2016;51(2):170-82.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888