Número Atual: Julho-Setembro 2020 - Volume 4 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
Síndrome de Hiper IgD: espectros clínicos, achados genéticos e condutas terapêuticas
Hyper-IgD syndrome: clinical spectrum, genetic findings, and therapeutic approaches
Alex Isidoro Ferreira Prado1,2; Fabio Fernandes Morato Castro1; Jorge Kalil1; Myrthes Toledo Barros1; Leonardo Oliveira Mendonça1,2
DOI: 10.5935/2526-5393.20200049
1. Universidade de São Paulo, Departamento de Imunologia Clínica e Alergia - São Paulo, SP, Brasil
2. Hospital 9 de Julho, Ambulatório de Doenças Raras - São Paulo, SP, Brasil
Endereço para correspondência:
Leonardo Oliveira Mendonça
E-mail: leonardo.oliveira.mendonca@gmail.com
Submetido em: 11/05/2020
Aceito em: 12/09/2020
Não foram declarados conflitos de interesse associados à publicação deste artigo
RESUMO
A deficiência de mevalonato quinase (MVK; MIM #142680; ORPHA #343) é uma doença genética, espectral, rara, associadas a mutações ao longo do gene MVK causando distúrbios na síntese do colesterol, que culminam em: inflamação sistêmica com febre, adenopatia, sintomas abdominais e outros achados clínicos. Enquanto no polo leve da doença os achados mais comuns são febres recorrentes com linfadenopatia, no polo mais grave adiciona-se o acometimento do sistema nervoso central (meningites assépticas, vasculites e atraso do desenvolvimento neuropsicomotor) e do sistema hematopoiético (síndrome de ativação macrofágica). Apesar de inúmeras terapêuticas, os bloqueadores da interleucina-1 ainda são os únicos medicamentos capazes de controlar a doença e de impedir a evolução para amiloidose. Os estudos atuais visam tentar novos tratamentos, como o transplante de células-tronco hematopoiéticas, ou mesmo a terapia gênica.
Descritores: Imunoglobulina D, deficiência de mevalonato quinase, doenças hereditárias autoinflamatórias.
INTRODUÇÃO
A deficiência de mevalonato quinase (DMQ - MIM #142680; ORPHA #343) ou síndrome de Hiper IgD (SHID) compõe um grupo de doenças espectrais inseridas no grupo das síndromes autoinflamatórias. Descrita pela primeira vez em 1984, por van der Meer e cols.1, como uma síndrome febril com hipergamaglobulinemia IgD, geneticamente é caracterizada por mutações ao longo do gene MVK responsável pela codificação da proteína mevalonato quinase. Sua herança é autossômica recessiva, e 80% dos pacientes carregam a mutação V377I. Apesar disso, a grande maioria dos pacientes com a doença são heterozigotos compostos2.
A proteína mevalonato quinase atua na via do metabolismo dos colesteróis e dos isoprenoides, tendo como função transformar o mevalonato, oriundo da molécula ß-hidroxi-ß-metilglutaril-CoA (HMG-CoA), em um isoprenoide que poderá ser convertido posteriormente para colesterol em nível celular ou adicionado a proteínas. Determinadas enzimas, como a farnesil transferase, são responsáveis pela prenilação de proteínas - adição de cadeias lipídicas isoprenoides nas proteínas, modificando sua conformidade e função3. Quando ocorre redução dos isoprenoides, um grupo pequeno de GTPases é afetado negativamente, incluindo Rac1 e RhoA. Em um estudo com células humanas viu-se que níveis reduzidos de RhoA por ausência de prenilação mantinha a atividade de GTP elevada com síntese de proteína quinase B e, por consequência, maior síntese de RNAm de interleucina 1B (IL1-b)2,4,5. Portanto, tais alterações demonstram que a enzima mevalonato quinase desempenha um importante papel no mecanismo da inflamação, e que sua ausência qualitativa ou quantitativa está associada à elevação de IL1β6. A Figura 1 traz um esquema representativo da fisiopatologia da formação da interleucina 1 derivada do gene MVK.
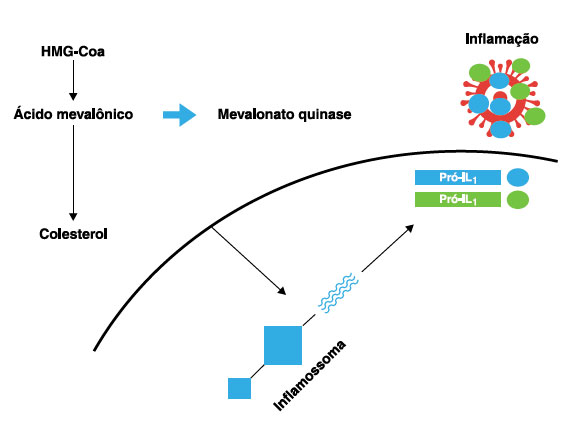
Figura 1
Esquema ilustrativo da ativação do inflamossoma intracelular a partir do defeito de metabolização da HMG-Coa da ausência de mevalonato quinase. O colesterol funciona como um DAMP (padrões moleculares associados a perigo) que culmina com a produção excessiva de IL1ß
Clinicamente, a DMQ possui dois polos principais que se relacionam com a gravidade e a atividade enzimática da MVK: em um polo de gravidade moderada, a síndrome de hiper IgD (SHID), no qual a atividade da MVK está entre 1% a 10% do normal, e a acidúria mevalônica (AM), polo mais grave em que a atividade da MVK está inferior a 1%7. A maioria dos casos descritos localiza-se na Europa Ocidental, com aproximadamente 60% ocorrendo em holandeses e franceses8. Ainda não há dados brasileiros sobre a prevalência da doença que, apesar de rara, deve ser subdiagnosticada em nível mundial.
Este trabalho propõe-se a revisar os principais achados clínicos, laboratoriais e genéticos, bem como as opções terapêuticas disponíveis.
MATERIAL E MÉTODOS
Foram feitas buscas nas redes de dados PubMed, Bireme e Cochrane, utilizando ou Hiper Igd (hyper igd ) ou Deficiência de mevalonato quinase (Mevalonate kinase deficiency ), sem uso de qualquer filtro. Os seguintes temas, isolados ou em conjunto, foram considerados na seleção dos artigos: diagnóstico clínico, diagnóstico laboratorial, diagnóstico genético, tratamento clínico, transplante de medula óssea, terapia gênica e prognóstico. Os resultados foram descritos de forma narrativa, com revisão crítica dos principais tópicos levantados.
RESULTADOS
Diagnóstico clínico
Até o momento desta publicação, a DMQ não apresenta critérios diagnósticos clínicos. Caracteristicamente, afeta igualmente ambos os sexos, mas seus sintomas e duração variam entre cada indivíduo e em cada espectro clínico, sendo que os sintomas mais graves apresentam-se na forma da AM. As manifestações imunológicas ocorrem como picos de inflamação sistêmica recorrentes com intervalos livres de sintomas. As manifestações clínicas principais são febre, acompanhada de surtos de adenopatia, faringites, dores abdominais, artrites e lesões cutâneas. Os desencadeantes desse processo autolimitado podem variar, mas sabe-se que a vacinação é um dos principais gatilhos conhecidos. Contudo, apesar da imunização ser identificada como 70% dos gatilhos nas DMQ, o procedimento mostra-se seguro nestes pacientes9-11.
A febre pode chegar a 40 ºC, com duração de 3 a 7 dias; tem início precoce, podendo manifestar-se nos primeiros dias após o nascimento, incluindo como diagnósticos diferenciais as sepses neonatais e, na grande maioria das vezes, os sintomas iniciam-se antes do primeiro ano de vida12. Estes episódios febris apresentam boa resposta quando tratados com corticoides e anti-inflamatórios.
A adenopatia é o segundo achado clínico mais comum, acometendo as cadeias cervicais, e admite diagnósticos diferenciais como as síndromes "monolike". As erupções cutâneas variam em sua apresentação, e se manifestam como exantema morbiliforme, máculo-papulares, urticariformes, eritema nodoso, placas similares às celulites, erupções purpúricas como Henoch-Schölein e até úlceras em mucosas oral e vaginal em 50% dos casos13.
A dor abdominal é uma das manifestações mais incapacitantes e de grande impacto na qualidade de vida. Acompanha muitas vezes diarreia, podendo confundir pela manifestação similar a uma doença inflamatória intestinal. Em alguns casos, os pacientes são submetidos a laparotomias, sem quaisquer achados importantes. Outro achado comum é a adenopatia mesentérica14.
As artralgias são vistas em períodos de crise, e mais raramente os pacientes podem apresentar artrite deformante de grandes articulações.
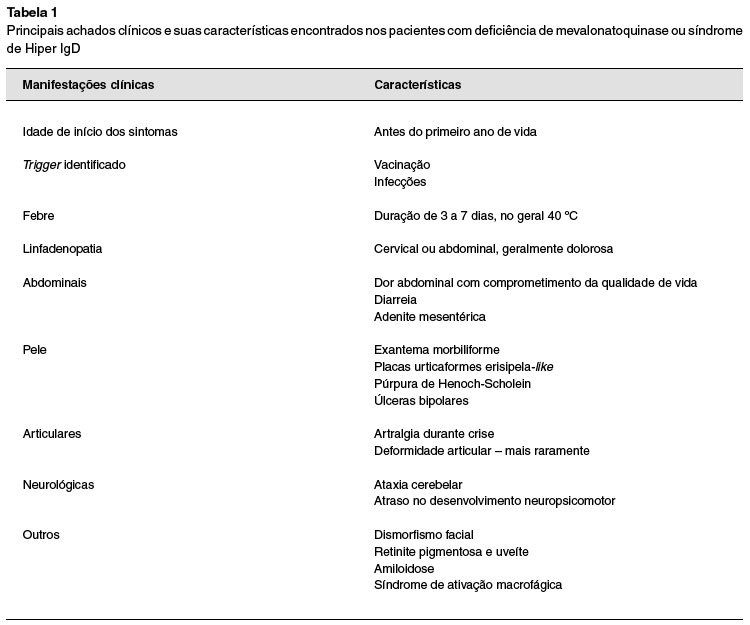
A AM apresenta as mesmas manifestações descritas acima, adicionando comprometimento neurológico grave. Pode ocorrer atraso no desenvolvimento, atrofia cerebelar, ataxia, dismorfismo facial e miopatia. Alguns casos na literatura também descrevem alterações oculares, como retinite pigmentosa, uveíte e esclera azul. Neste polo também é possível a evolução para amiloidose (em menos de 3% dos casos) e complicações graves, como síndrome de ativação macrofágica2,13,15. A Tabela 1 resume todos os achados clínicos encontrados na DMQ.
Diagnóstico laboratorial
Alterações laboratoriais gerais e específicas
A análise laboratorial do paciente com DMQ independe do seu polo clínico, e deve ser feita durante e fora do momento da crise, como na maioria das doenças autoinflamatórias. No momento da crise febril, ocorre elevação de polimorfonucleares (leucocitose com neutrofilia) e aumento das provas inflamatórias que, no geral, apresentam-se normais fora dos períodos de crise. Durante a atividade de doença é possível constatar o aumento de marcadores inflamatórios de fase aguda, como velocidade de hemossedimentação (VHS), proteina C-reativa (PCR), ferritina e substância amiloide A (SAA), que devem idealmente ser dosados também em momentos nos quais o paciente encontra-se assintomático16. A dosagem das imunoglobulinas demonstra a elevação policlonal de IgA, IgD, e até IgM, auxiliando na investigação. Entretanto, em 10% dos casos o valor de IgD pode ser normal, e sua medição sérica isolada não confirma o diagnóstico na ausência da mutação. Além disso, o aumento desta imunoglobulina pode ser vista em outras síndromes, como a síndrome de febre periódica, estomatite aftosa, faringite e adenite cervical (PFAPA)17.
A dosagem sérica de colesterol não é de grande valia, mas pode demonstrar valores reduzidos durante crises; por outro lado, a dosagem do ácido mevalônico urinário traz grande especificidade em pacientes no polo mais grave da doença (AM), e auxilia como triagem de pacientes candidatos ao estudo genético com 90% de especificidade2.
Alterações histopatológicas
Apesar da constelação de manifestações dermatológicas, a biopsia de pele habitualmente não traz alterações específicas que permitam diferenciar a DMQ de outras doenças autoinflamatórias, nem segregar os polos da doença. Estudos dermatológicos demonstram como principais achados histopatológicos a presença de dermatose neutrofílica, figuras de leucocitoclasia e alguns histiócitos18.
Diagnóstico genético
O gene MVK localiza-se no cromossomo 12 (12q24), e com os estudos atuais seu sequenciamento permitiu identificar uma série de mutações, com relevância clínica na DMQ. Mais de 120 variantes já foram encontradas, sendo em sua maioria de polimorfismos de nucleotídeo único que com mutações missense prejudicam a estabilidade da enzima MVK. Apesar de a herança ser autossômica recessiva, a maioria dos pacientes carreia mutação bialélica no gene MKV, e poucos deles têm apenas uma mutação clínica identificada, mas clinicamente definidos. Mutações que trazem alterações no núcleo da enzima gerada se associam mais frequentemente ao espectro de maior gravidade, provavelmente por instabilidade na estrutura quartenária da MVK, e mutações em substituição de terminal C, como a V377I, geralmente estão associadas ao outro polo da doença (HIDS), e raramente apresentam clínica de AM2. A lista com todas as variantes já encontradas ao longo dos 11 exons e introns do gene MVK encontra-se disponível na Internet através do link: https://infevers.umai-montpellier.fr/web/.
Principais achados clínicos e suas características encontrados nos pacientes com deficiência de mevalonatoquinase ou síndrome de Hiper IgD
Tratamento clínico
Antes da era dos biológicos, os pacientes portadores da doença controlavam suas crises com o uso de anti-inflamatórios não esteroidais, ou mesmo corticoterapia. Apesar da resposta satisfatória em alguns casos, os pacientes que estão no polo mais grave da doença permanecem enfrentando grandes desafios pela condição gerada principalmente pelas alterações neurológicas da AM. O corticoide pode ser administrado durante as crises na dose de 1 mg/ kg, trazendo melhora dos sintomas aos pacientes19. A DMQ não apresenta boa resposta ao uso da colchicina, pouco controle das febres e das crises abdominais, trazendo mais efeitos adversos da medicação do que melhora da qualidade de vida. O uso das estatinas já foi aventado, entretanto, em alguns estudos notou-se piora clínica e desencadeamento das crises com sua administração, sendo, desta forma, abandonada como opção terapêutica20. Com o advento dos monoclonais, os pacientes experimentaram melhor resposta no controle da doença.
O uso de etanercept e adalimumab (anticorpos monoclonais anti-TNF) são considerados primeira linha terapêutica, porém estão associados a resposta parcial2. O uso de anti-IL1 é mais eficaz, uma vez que envolve as bases fisiopatológicas da doença. O anankira (antagonista recombinante do receptor de IL-1) vem sendo utilizado de diversas formas, sob demanda ou em doses fixas, apresentando melhor resposta nesta última opção21. Já o canaquinumabe (anticorpo monoclonal humano bloqueador da IL-1β) demonstrou nos últimos estudos resposta completa em até 50% dos pacientes com menores efeitos adversos, quando comparado ao anankira22-24. Pode ser considerado ainda o uso de tocilizumab (anticorpo monoclonal direcionado contra o receptor da IL-6) apesar de poucos estudos mostrando efetividade25.
Estudos realizados em 2011, após o adequado conhecimento das vias metabólicas do colesterol, permitiram testes com drogas conhecidas como inibidores da farnesiltransferase - tipifarnib e lonafarnib. Tais medicações aumentaram os níveis de colesterol e redistribuíram os compostos intermediários da via do mevalonato quinase com redução da regulação positiva da IL-1β26. Ainda menos convincentes, alguns estudos tentaram demonstrar o uso do alendronato no tratamento da DMQ pela melhora clínica em um dos pacientes portadores da doença, e que também apresentava doenças ósseas; entretanto sua vantagem não foi comprovada in vitro, mimetizando inclusive alterações bioquímicas vistas na doença em si27,28.
O transplante de células-tronco hematopoéticas (TCTH) pode ser indicado para casos mais graves e refratários à terapia biológica habitual. Em todos os relatos de TCTH, as indicações foram quadro clínico de espectro mais grave, principalmente quadros neurológicos e gastrointestinais refratários tanto ao uso de corticoide sistêmico em doses elevadas, anti-TNF alfa e anti-IL129,30.
A terapia gênica, agora em estudo para as imunodeficiências, poderia ser uma alternativa futura para a doença, considerando seus mecanismos e o conhecimento atual sobre as alterações no gene MVK. Entretanto, são necessários estudos que possam relacionar as doenças autoinflamatórias e o seu tratamento por meio da genética.
Um fluxograma contendo o diagnóstico e tratamento da DMQ está ilustrado na Figura 2.
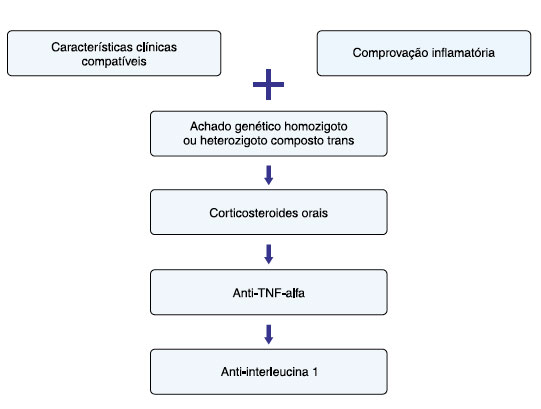
Figura 2
Fluxograma para diagnóstico e tratamento das síndromes associadas a deficiência de mevalonato quinase
Aconselhamento genético
Por se tratar de uma doença genética, é de responsabilidade ética do médico que acompanha o paciente realizar o aconselhamento genético, que deve ser feito em conjunto com médico geneticista. Este procedimento consiste em orientar a família sobre a chance de outra prole herdar a mutação, e resultar em outro membro afetado na família. Deve-se levar em consideração o tipo de mutação encontrada e a forma de herança desta mutação.
Prognóstico
Os pacientes portadores da doença que estão no polo de menor gravidade passam por períodos assintomáticos, reduzindo crises ao longo da adolescência até a idade adulta. Já pacientes portadores da AM terão comprometimento importante da qualidade de vida pelo acúmulo progressivo de alterações neurológicas ao longo da vida. Quando comparado com outras síndromes autoinflamatórias, a DMQ tem sua evolução para amiloidose mais baixa (3% ou menos)1,2. Vale ressaltar a importância do acompanhamento clínico, bem como dos exames laboratoriais do paciente com dosagens de provas inflamatórias durante e fora das crises. Pacientes com elevação constante da SAA devem ser rastreados com biopsia de gordura periumbilical, utilizando o método de coloração com vermelho do congo para o diagnóstico tecidual. Uma vez confirmado este diagnóstico, o paciente terá comprometimentos além dos conhecidos da DMQ, como insuficiência renal e alterações hepáticas32.
CONCLUSÃO
A síndrome de hiper IgD ou deficiência de mevalonato quinase é uma doença rara, espectral que deve ser lembrada como diagnóstico diferencial dentre as síndromes febris recorrentes, as linfadenopatias benignas, as doenças inflamatórias intestinais de início precoce, as doenças neurológicas inflamatórias da infância e as síndromes de ativação macrofágicas. Os achados laboratoriais devem confirmar a inflamação sistêmica, mas são inespecíficos. O achados de elevação sérica de imunoglobulina D e de acidúria mevalônica na urina são sugestivos, mas não exclusivos da síndrome. O diagnóstico final é através dos achados genéticos de mutações bialélicas no gene MVK, e também têm importante função na orientação genética familiar. Até este momento, o único tratamento eficaz é através do bloqueio de interleucina 1.
REFERÊNCIAS
1. van der Meer JW, Vossen JM, Radl J, van Nieuwkoop JA, Meyer CJ, Lobatto S, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet. 1984;1(8386):1087-90.
2. Favier LA, Schulert GS. Mevalonate kinase deficiency: current perspectives. Appl Clin Genet. 2016;9:101-10.
3. Gabor KA, Fessler MB. Roles of the mevalonate pathway and cholesterol trafficking in pulmonary host defense. Curr Mol Pharmacol. 2017;10(1):27-45.
4. van der Burgh R, Pervolaraki K, Turkenburg M, Waterham HR, Frenkel J, Boes M. Un-prenylated RhoA contributes to IL-1beta hypersecretion in mevalonate kinase deficiency model through stimulation of Rac1 activity. J Biol Chem. 2014;289(40):27757-65.
5. Hoffmann GF, Charpentier C, Mayatepek E, Mancini J, Leichsenring M, Gibson KM, et al. Clinical and biochemical phenotype in 11 patients with mevalonic aciduria. Pediatrics. 1993;91(5):915-21.
6. van der Burgh R, Ter Haar NM, Boes ML, Frenkel J. Mevalonate kinase deficiency, a metabolic autoinflammatory disease. Clin Immunol. 2013;147(3):197-206.
7. Mandey SHL, Schneiders MS, Koster J, Waterham HR. Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency. Human Mutation. 2006;27(8):796-802.
8. Zhang S. Natural history of mevalonate kinase deficiency: a literature review. Pediatr Rheumatol Online J. 2016;14(1):30.
9. Haraldsson A, Weemaes CM, De Boer AW, Bakkeren JA, Stoelinga GB. Immunological studies in the hyper-immunoglobulin D syndrome. J Clin Immunol. 1992;12(6):424-8.
10. van der Hilst JC, Bodar EJ, Barron KS, Frenkel J, Drenth JP, van der Meer JW, et al. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore). 2008;87(6):301-10.
11. Signa S, Cerinic CM, Toniolo E, Bustaffa M, D'alessandro M, Volp S, et al. OP0260 Vaccination safety and coverage in an Italian cohort of autoinflammatory diseases. Ann Rheum Dis. 2019;78:211.
12. Padeh S, Berkun Y. Periodic fever syndromes. In: Shoenfeld Y, Cervera , Gershwin ME (eds.) Diagnostic Criteria in Autoimmune Diseases. Humana Press; 2008. p. 201-7.
13. Havnaer A, Han G. Autoinflammatory Disorders: A Review and Update on Pathogenesis and Treatment. Am J Clin Dermatol. 2019;20(4):539-64.
14. Pieri C, Insalaco A, Taddio A, Barbi E, Lepore L, Tommasini A, et al. Mevalonate kinase deficiency: Various aspects of the same disease. Medico e Bambino. 2013;32:501-6.
15. Pieri C, Taddio A, Insalaco A, Barbi E, Lepore L, Ventura A, et al. Different presentations of mevalonate kinase deficiency: A case series. Clin Exp Rheumatol. 2015;33:437-42.
16. Kastner DL. Hereditary periodic fever syndromes. Hematology. 2005;2005(1):74-81.
17. Ammouri W, Cuisset L, Rouaghe S, Rolland M-O, Delpech M, Grateau G, et al. Diagnostic value of serum immunoglobulinaemia D level in patients with a clinical suspicion of hyper IgD syndrome. Rheumatology. 2007;46(10):1597-600.
18. Pace S, Bingham J, Royer M. Histopathologic features in a case of hyperimmunoglobulinemia D syndrome. Indian Dermatol Online J. 2015;6(Suppl 1):S33-S6.
19. ter Haar N, Lachmann H, Özen S, Woo P, Uziel Y, Modesto C, et al. Treatment of auto-inflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis. 2013;72(5):678-85.
20. Durel C-A, Aouba A, Bienvenu B, Deshayes S, Coppéré B, Gombert B, et al. Observa-tional Study of a French and Belgian Multicenter Cohort of 23 Patients Diagnosed in Adulthood With Mevalonate Kinase Deficiency. Medicine (Baltimore). 2016;95(11):e3027-e.
21. Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis. 2011;70(12):2155-8.
22. Rossi-Semerano L, Fautrel B, Wendling D, Hachulla E, Galeotti C, Semerano L, et al. Tolerance and efficacy of off-label antiinterleukin- 1 treatments in France: a nationwide survey. Orphanet J Rare Dis. 2015;10:19.
23. De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N Engl J Med. 2018;378(20):1908-19.
24. ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recom-mendations for the management of autoinflammatory diseases. Ann Rheum Dis. 2015;74(9):1636-44.
25. Rafiq NK, Lachmann H, Joensen F, Herlin T, Brogan PA. Tocilizumab for the Treatment of Mevalonate Kinase Deficiency. Case Rep Pediatr. 2018;2018:3514645.
26. De Leo L, Marcuzzi A, Decorti G, Tommasini A, Crovella S, Pontillo A. Targeting farnesyl-transferase as a novel therapeutic strategy for mevalonate kinase deficiency: In vitro and in vivo approaches. Pharmacol Res. 2010;61(6):506-10.
27. Tricarico PM, Girardelli M, Kleiner G, Knowles A, Valencic E, Crovella S, et al. Alendro-nate, a double-edged sword acting in the mevalonate pathway. Mol Med Rep. 2015;12(3):4238-42.
28. Cantarini L, Vitale A, Magnotti F, Lucherini OM, Caso F, Frediani B, et al. Weekly oral alendronate in mevalonate kinase deficiency. Orphanet J Rare Dis. 2013;8:196.
29. Neven B, Valayannopoulos V, Quartier P, Blanche S, Prieur AM, Debré M, et al. Allogeneic Bone Marrow Transplantation in Mevalonic Aciduria. N Engl J Med. 2007;356(26):2700-3.
30. Chaudhury S, Hormaza L, Mohammad S, Lokar J, Ekong U, Alonso EM, et al. Liver Transplantation Followed by Allogeneic Hematopoietic Stem Cell Transplantation for Atypical Mevalonic Aciduria. Am J Transplant. 2012;12(6):1627-31.
31. Giardino S, Lanino E, Morreale G, Madeo A, Di Rocco M, Gattorno M, et al. Long-term outcome of a successful cord blood stem cell transplant in mevalonate kinase deficiency. Pediatrics. 2015;135(1):e211-5.
32. Li Cavoli G, Passantino D, Tortorici C, Bono L, Ferrantelli A, Giammarresi C, et al. Renal amyloidosis due to hyper-IgD syndrome. Nefrologia. 2012;32(6):865-6.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888