Número Atual: Janeiro-Março 2020 - Volume 4 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo Original
Erros inatos de imunidade: tempo de diagnóstico e episódios infecciosos em pacientes ambulatoriais
Inborn errors of immunity: time to diagnosis and infections in outpatients
Naiara de Oliveira Pazian1; Laura Lúcia Cogo2; Denise Eli2; Carlos Antônio Riedi3; Herberto Jose Chong-Neto3; Nelson Augusto Rosario-Filho4
1. Universidade Federal do Paraná, Farmacêutica-Bioquímica do Complexo Hospital de Clínicas - Curitiba, PR, Brasil
2. Universidade Federal do Paraná, Unidade de Laboratório de Análises Clínicas - Curitiba, PR, Brasil
3. Universidade Federal do Paraná, Professor Adjunto de Pediatria - Curitiba - Paraná - Brasil
4. Universidade Federal do Paraná, Professor Titular de Pediatria - Curitiba - Paraná - Brasil
Endereço para correspondência:
Naiara de Oliveira Pazian
E-mail: naiarapazian@hotmail.com
Submetido em: 21/02/2020
Aceito em: 27/02/2020
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
INTRODUÇÃO: Os erros inatos de imunidade (EII) são distúrbios que ocasionam danos no desenvolvimento e/ou função do sistema imunológico. O diagnóstico muitas vezes não é realizado de imediato devido ao pouco conhecimento sobre as doenças, que leva a complicações graves e diminui a sobrevida e qualidade de vida desses pacientes. O objetivo desse estudo foi avaliar o tempo para o diagnóstico e as ocorrências infecciosas que acometeram pacientes com EII no decorrer de sua vida até o momento do diagnóstico.
MÉTODO: Foi realizado um estudo transversal, retrospectivo, em pacientes atendidos pelo serviço de Alergia, Imunologia e Pneumologia do Complexo Hospital de Clínicas da Universidade Federal do Paraná (CHC-UFPR), no período de junho de 1993 a março de 2019. Foram excluídos pacientes sem história prévia ao diagnóstico e com diagnóstico não confirmado de EII ou indefinido.
RESULTADOS: Dos 57 pacientes incluídos no estudo, a maioria (n = 34) era do sexo masculino. A idade ao diagnóstico variou de 2 até 38 anos, sendo a média 9 anos. Dentre as imunodeficiências, 43 (75,4%) tinham deficiência de anticorpos, 10 (17,5%) deficiência combinada, 3 (5,3%) deficiência de fagócitos e 1 (1,8%) deficiência de complemento. Em relação às infecções, os pacientes apresentaram mais de um episódio infeccioso, e também sofreram acometimento em mais de um sítio anatômico. As infecções mais frequentes foram as do trato respiratório inferior (80,7%), seguido das infecções do trato respiratório superior (50,9%). Foi encontrado um atraso médio de diagnóstico de 66,1 meses, sendo que 10,5% dos pacientes foram a óbito.
CONCLUSÃO: Apesar de já serem bem caracterizados, os EII ainda possuem diagnóstico tardio, levando os pacientes a complicações graves, e até à morte.
Descritores: Imunidade, infecção, diagnóstico.
INTRODUÇÃO
Os erros inatos de imunidade (EII) são um grupo heterogêneo de doenças de origem genética, que estão associadas a defeitos congênitos relacionadas a danos no desenvolvimento e/ou função do sistema imunológico1. Este grupo de doenças pode acometer tanto a imunidade adaptativa, que corresponde à resposta imunológica humoral e celular, como a imunidade inata, que são mecanismos inespecíficos mediados por barreiras epiteliais, proteínas do sistema complemento, distúrbios de células T e células B, entre outros2.
Os EII, são classificados em oito categorias definidas pela International Union of Immunological Societies (IUIS): imunodeficiências combinadas das células T e B, síndromes de imunodeficiência bem definidas ou imunodeficiências combinadas com características sindrômicas, as deficiências predominantemente de anticorpos, doenças da desregulação imunológica, defeitos de número ou função de fagócitos, defeitos na imunidade inata, distúrbios autoinflamatórios e deficiência de complemento3.
A distribuição e prevalência das EII é bastante variada entre as populações, e ainda é considerada uma doença rara. No entanto, ela acomete cerca de 1 a 2% da população norte-americana, sendo que já foram classificados cerca de 440 distúrbios específicos. No Brasil, a real frequência ou distribuição na população ainda é desconhecida4.
As manifestações mais comuns das EII são as infecções de repetição, que variam conforme a porção acometida do sistema imunológico, tendo variações de infecções de risco leve a grave, incluindo até mesmo infecções sistêmicas5,6. Comumente essas infecções estão associadas com complicações, ocorrência de infecções de múltiplos sítios anatômicos, frequentemente com resistência ao tratamento convencional ou que tenham como causa microrganismos oportunistas7.
Apesar da maioria dos casos de EII ser diagnosticada ainda quando criança, é comum o diagnóstico em jovens e adultos. No entanto, os sinais clínicos diferem entre essas duas populações. Os sinais e sintomas encontrados em pacientes pediátricos progridem rapidamente. Já na maioria dos adultos, os pacientes possuem manifestações de leve a moderada. Por esse motivo, é fundamental que se tenha um histórico das principais manifestações para o auxílio ao diagnóstico8.
O objetivo deste estudo foi avaliar o tempo para o diagnóstico e as ocorrências infecciosas que acometeram pacientes com EII no decorrer de sua vida até o momento do diagnóstico.
MÉTODOS
Foi realizado um estudo transversal e retrospectivo de 57 pacientes com diagnóstico de EII, acompanhados no serviço de Alergia e Imunologia Pediátrica do Complexo Hospital de Clínicas da Universidade Federal do Paraná (CHC-UFPR), que foram atendidos pelo setor a partir de junho de 1993 até o período de março de 2019. O estudo foi aprovado pelo Comitê de Ética da instituição com o parecer n° 02457718.5.0000.0096. Foram excluídos do estudo indivíduos sem diagnóstico definido, ou quando o diagnóstico de EII foi descartado, e aqueles que não possuíam história prévia ao diagnóstico no prontuário.
Os pacientes foram organizados em grupos conforme as categorias das EII, correlacionando com os sítios infecciosos acometidos. Foram analisados também idade, sexo, início das manifestações clínicas, tempo de diagnóstico e óbito. Os dados foram tabulados no software Microsoft Office Excel e avaliados usando média, medianas e porcentagens.
RESULTADOS
Dos 57 pacientes analisados, 59,6% (n = 34) eram do gênero masculino e 40,4% (n = 23) do gênero feminino, obtendo uma razão homem:mulher de 1,5:1. Essa distribuição variou nas doenças ligadas ao cromossoma X, na qual obteve uma razão de 3,2:1. Em relação à faixa etária, foram classificados conforme a idade que apresentavam no momento do diagnóstico. Dessa forma, a idade de diagnóstico variou de 2 até 38 anos, a média 9 anos (Figura 1).
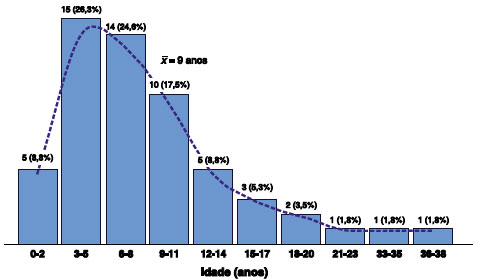
Figura 1 Distribuição dos pacientes com erros inatos de imunidade por faixa etária ao diagnóstico
Dentre as oito categorias definidas pela IUIS, a deficiência predominantemente de anticorpos foi a mais encontrada, sendo diagnosticada em 75,4% da população em estudo, na qual a deficiência específica de anticorpos anti-pneumococos (28%) e a deficiência de IgA (16,8%) foram as mais frequentes nesta categoria. Em seguida, as imunodeficiências combinadas com características associadas ou sindrômicas (17,5%), defeitos congênitos no número e/ou função de fagócitos (5,3%) e, por último, as deficiências do sistema complemento, sendo apenas 1 paciente diagnosticado (1,8%).
As infecções encontradas foram subdivididas conforme a categoria de imunodeficiência e de acordo com o sítio de acometimento, em que foi observado a frequência das infecções, representada na Tabela 1. Vale ressaltar que cada paciente apresentou vários episódios infecciosos e também sofreu acometimento em mais de um sítio anatômico.

O início das manifestações infecciosas foi em média de 41,4 meses, com mediana de 12 meses (1-240 meses). Com esses dados, foi possível estimar o atraso médio do diagnóstico, o qual variou de acordo com a categoria de imunodeficiência (Figura 2).
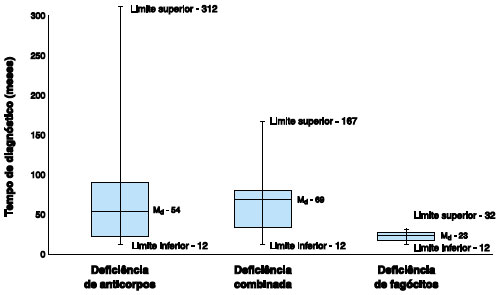
Figura 2 Tempo de diagnóstico dos erros inatos de imunidade (em meses)
Dos 57 pacientes, 10,5% foram a óbito por complicações infecciosas, sendo 4 pacientes (66,7%) classificados como imunodeficiência combinada, e 2 pacientes (33,3%) como deficiência de anticorpos.
DISCUSSÃO
As EII representam um problema de saúde devido a grande diversidade de aspectos clínicos e defeitos genéticos, tornando um desafio o seu reconhecimento e o diagnóstico nos pacientes9.
O ambulatório de Alergia e Imunologia Pediátrica do CHC-UFPR foi o primeiro do Brasil a receber o título de Centro de Excelência pela World Allergy Organization (WAO), e por esse motivo é rotineiro que pacientes com suspeita de EII de outras regiões sejam encaminhados para o serviço. Além disso, setores de outras especialidades dentro do próprio hospital acabam realizando o encaminhamento quando há uma suspeita, assim como pacientes com apresentações mais graves são encaminhados às demais especialidades.
Ao avaliarmos a faixa etária de diagnóstico, observou-se uma média acima da maioria dos estudos10-12. Pesquisa realizada em centro de referência no Brasil, mostrou um número elevado de pacientes com diagnóstico em idade acima da esperada, tendo 38% dos pacientes idade entre 5-20 anos, e 35% com idade superior a 20 anos5. A idade avançada ao diagnóstico pode estar relacionada à falta de conscientização da população, bem como dos profissionais de saúde sobre os sinais de alerta13 para que ocorra a procura pela atenção primária, assim como o encaminhamento precoce à atenção terciária.
Na distribuição por gênero, assim como nosso centro, vários estudos demonstraram predomínio do gênero masculino11,14. Este fato pode estar relacionado à algumas imunodeficiências serem desencadeadas por mutações em genes presentes no cromossoma X, o que aumenta a predisposição da ocorrência em pacientes do gênero masculino15.
Em relação à distribuição dos EII, as imunodeficiências humorais foram as mais encontradas, assim como em outros estudos5,16. No entanto, a maior população que representou esse grupo foi a de deficiências específicas de anticorpos anti-pneumococos, que afeta a resposta da IgG aos antígenos polissacarídeos do pneumococo e resulta em infecções bacterianas recorrentes16,17. O predomínio dessa deficiência, favoreceu o desenvolvimento de infecções sinopulmonares, que levaram as infecções de trato respiratório serem as mais frequentes na população em estudo17-19. As deficiências de IgA são as mais relatadas na América Latina, discordando dos resultados encontrados, a qual deve estar relacionada às manifestações leves dessa categoria ou por serem assintomáticas e pacientes não são encaminhados ao serviço especializado3,20.
O atraso no diagnóstico foi estimado a partir do início dos sintomas infecciosos, relatados por um acompanhante ou pelo próprio paciente, até o momento que foi confirmado o diagnóstico de EII. Dessa forma, encontramos atraso médio de 66,1 meses, consideravelmente mais elevado do que nos estudos realizados em outros países, como Peru21, Omã12, México10 e Arábia Saudita22, que descreveram atraso médio de diagnóstico de 12,2 meses, 21 meses , 22 meses e 39 meses, respectivamente. A amostragem em nosso estudo foi composta principalmente por pacientes com diagnóstico de deficiência de anticorpos, normalmente com manifestações mais leves, e por esse motivo apresentam retardo no diagnóstico. Para que ocorra diagnóstico precoce é fundamental a disseminação sobre os sinais de alerta da doença, tanto para a população como para os profissionais de saúde, para que, desta forma, propiciem o encaminhamento ao serviço especializado o mais rápido possível. O resultado reflete a demora no processo, já que a partir da primeira consulta no CHC-UFPR foi obtido um tempo de diagnóstico semelhante aos outros centros.
Ao analisarmos a taxa de mortalidade, encontramos valores semelhantes ao estudo realizado por Lim et. al., de 7,1% de óbitos23. Porém, a taxa de mortalidade é inferior quando comparada a alguns centros de referências, podendo variar de 18% a 34,5%11,12. Isso pode ser justificado tanto pela representação amostral de cada estudo, quanto pelo fato da nossa população não conter pacientes com imunodeficiência combinada severa (SCID), que normalmente são encaminhados para transplante de medula óssea, e possuem maior chance de complicações e óbitos24.
CONCLUSÃO
Os EII, apesar de bem caracterizados e documentados, representam um desafio em seu diagnóstico e tratamento. A diversidade das categorias da doença, as diferentes deficiências e mutações e até mesmo a infecção como manifestação mais frequente, faz com que dificulte o reconhecimento pelos profissionais de saúde. A suspeita através dos sinais de alerta no atendimento primário é de extrema importância para que os casos sejam encaminhados ao serviço especializado antes mesmo de manifestações mais graves e irreversíveis, oferecendo um melhor prognóstico e aumentando a qualidade de vida desses pacientes.
REFERÊNCIAS
1. Yu JE, Orange JS, Demirdag YY. New primary immunodeficiency diseases: context and future. Curr Opin Pediatr. 2018;30(6):806-20.
2. Routes J, Abinum M, Al-Herz W, Bustamante J, Condino NA, Morena MT, et al. ICON: The early diagnosis of congenital immunodeficiencies. J Clin Immunol. 2014 May;34(4):398-424.
3. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: 2019. Update on the classification from the international union of immunological societies expert committee. J Clin Immunol. 2020 Jan;40(1):24-64. doi: 10.1007/s10875-019-00737-x. Epub 2020 Jan 17.
4. Modell V, Quinn J, Ginsberg G, Gladue R, Orange J, Modell F. Modeling strategy to identify patients with primary immunodeficiency utilizing risk management and outcome measurement. Immunologic Res. 2017;65(3):713-20.
5. Carneiro-Sampaio M, Moraes-Vasconcelos D, Kokron CM, Jacob CM, Toledo-Barros M, Dorna MB, et al. Primary immunodeficiency diseases in different age groups: a report on 1,008 cases from a single Brazilian reference center. J Clin Immunol. 2013;33(4):716-24.
6. Bonilla FA, Bernstein IL, Khan DA, Ballas ZK, Chinen J, Frank MM, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;136(5):94:S1-63.
7. Abolhassani H, Rezaei N, Mohammadinejad P, Mirminachi B, Hammarstrom L, Aghamohammadi A. Important differences in the diagnostic spectrum of primary immunodeficiency in adults versus children. Expert Review of Clinical Immunology (Irã). 2015;11(2):289-302.
8. Aghamohammadi A, Mohammadinejad P, Abolhassani H, Rezaei N. The approach to children with recurrent infections. Iran J Allergy Asthma Immunol. 2012;11(2):89-109.
9. Dantas EO, Aranda CS, Rêgo Silva AM, Tavares FS, Severo Ferreira JF, Quadros-Coelho MA, et al. Doctors' awareness concerning primary immunodeficiencies in Brazil. Allergol Immunopathol. 2015;43(3):272-8.
10. Lugo R, Ramirez-Vazquez, Cruz H, Medina-Torres, Ramirez- Lopez, España-Cabrera, et al. Clinical features, non-infectious manifestations and survival analysis of 161 children with primary immunodeficiency in Mexico: a single center experience over two decades. J Clin Immunol. 2016;36(1):56-65.
11. Mellouli F, Mustapha IB, Khaled MB, Besbes H, Ouederni M, Mekki N, et al. Report of the Tunisian Registry of Primary Immunodeficiencies: 25-Years of Experience (1988-2012). J Clin Immunol. 2015;35(8):745-53.
12. Al-Tamemi S, Naseem SU, Al-Siyabi N, El-Nour I, Al-Rawas A, Dennison D. Primary Immunodeficiency Diseases in Oman: 10-Year Experience in a Tertiary Care Hospital. J Clin Immunol. 2016;36(8):785-92.
13. BRAGID. Imunodeficiência Primária. Os 10 sinais de alerta. [Internet] Disponível em: http://www.bragid.org.br/_download/10sinais.pdf. Acessado em: outubro/2019.
14. Halioui-Louhaichi S, Azzabi O, Mattoussi N, Labiadh H, Bousseta K, Tebib N, et al. Primary immunodeficiencies: Report of 33 Pediatric Tunisian cases. Tunis Med. 2016;94(4):320-5.
15. Winkelstein JA, Fearon E. Carrier detection of the X-linked primary immunodeficiency diseases using X-chromosome inactivation analysis. J Allergy Clin Immunol. 1990;85(6):1090-7.
16. Moleta FB, Chong-Silva DC, Riedi CA, Neto HJC, Rosário NA. Warning signs application in primary immunodeficiencies diagnosis. Jornal Paranaense de Pediatria. 2019;19(4):76-9.
17. Forte WCN, Konichi RYL, Sousa FM, Mosca T, Rego AM, Goudouris ES. Deficiência de anticorpos específicos antipolissacarídeos. Arq Asma Alerg Imunol. 2019;3(2):111-22.
18. Barreto BAP, Sarinho ESC, Stefani GP, Neto HJC, Chiabai J, Alonso MLO, et al. Deficiência específica de anticorpo antipolissacarídeo de pneumococo e resposta humoral a vacinas pneumocócicas: atualização em diagnóstico. Braz J Allergy Immunol. 2013;1(5):253-60.
19. Roxo JP. Primary immunodeficiency disease: Relevant aspects for pneumologists. Journal Bras Pneumology (São Paulo). 2014;35(10):1008-17.
20. Leiva LE, Zelazco M, Oleastro M, Carneiro-Sampaio M, Condino-Neto A, Costa-Carvalho BT, et al. Latin American Group for Primary Immunodeficiency Diseases. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J Clin Immunol. 2007;27(1):101-8.
21. Veramendi-Espinoza LE, Zafra-Tanaka JH, Pérez-Casquino GA, Córdova-Calderón WO. Diagnostic Delay of Primary Immunodeficiencies at a Tertiary Care Hospital in Peru- Brief Report. J Clin Immunol. 2017;37(4):383-7.
22. Al-Saud B, Al-Mousa H, Al Gazlan S, Al-Ghonaium A, Arnaout R, Al-Seraihy A, et al. Primary Immunodeficiency Diseases in Saudi Arabia: a Tertiary Care Hospital Experience over a Period of Three Years (2010-2013). J Clin Immunol. 2015;35(7):651-60.
23. Lim DL, Thong BY, Ho LP, Shek PC, Lou J, Leong KP, et al. Primary Immunodeficiency Diseases in Singapore - the Last 11 Years. Singapore Med J. 2003 Nov;44(11):579-86.
24. Fernandes JF, Kerbauy FR , Ribeiro AAF , Kutner JM , Camargo LFA, Stape A, et al. Allogeneic hematopoietic stem cell transplantation in children with primary immunodeficiencies: Hospital Israelita Albert Einstein experience. Einstein. 2011;9(2):140-4.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888