Número Atual: Janeiro-Fevereiro 2013 - Volume 1 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
Neutropenia congênita
Congenital neutropenia
Paolo Ruggero Errante1; Josias Brito Frazao2; Antônio Condino Neto3
DOI: 10.5935/2318-5015.20130006
1. VMD, PhD. Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de Sao Paulo (USP), SP
2. BSc, MSc. Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de Sao Paulo (USP), SP
3. MD, PhD. Departamento de Imunologia, Instituto de Ciências Biomédicas, Universidade de Sao Paulo (USP), SP
Endereço para correspondência:
Antônio Condino Neto
E-mail: condino@icb.usp.br
Submetido em 30.07.2012.
Aceito em 23.03.2013.
Agências financiadoras: CAPES/CNPq, FAPESP.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
Buscamos aqui revisar os mecanismos imunopatológicos relacionados à neutropenia congênita. O termo neutropenia congênita é utilizado para designar uma série de distúrbios neutropênicos, de caráter permanente, intermitente, grave (< 500 neutrófilos/mm3 de sangue), ou moderado (entre 500-1.500 neutrófilos/mm3 de sangue), que podem acometer pele e mucosa do trato respiratório e gastrintestinal. Quando a neutropenia é diagnosticada, ela deve ser distinguida das formas adquiridas, incluindo a neutropenia pós-viral e a autoimune, da forma congênita, que pode ser uma enfermidade isolada ou fazer parte de uma doença genética. Cinquenta por cento das formas congênitas de neutropenia apresentam manifestaçao extra-hematopoiética com resposta imune adaptativa normal e infecçoes recorrentes no início da vida. O tratamento destes pacientes tem por objetivo o controle e a prevençao de infecçoes através do uso profilático de antibióticos, e outra forma de tratamento consiste na utilizaçao de fator estimulador de colônia de granulócitos recombinante humano (rHUG-CSF), que aumenta o número de granulócitos, diminui o número infecçoes e melhora de forma significativa a sobrevida e qualidade de vida. A revisao foi realizada por levantamento bibliográfico de banco de dados obtidos através de pesquisa direta, LILACS, MEDLINE e capítulos de livros. A revisao literária demonstra a importância dos neutrófilos pela defesa do hospedeiro contra micro-organismos, e defeitos genéticos que envolvem estas células acarretam maior susceptibilidade a infecçoes microbianas em locais como pele e mucosa do trato respiratório e gastrintestinal. Estes defeitos genéticos dos neutrófilos envolvem o seu número, funçao, ou ambos. Como estes defeitos envolvendo fagócitos sao de caráter congênito e hereditário, as crianças sao os pacientes predominantes. Os neutrófilos apresentam um papel importante na imunidade inata, prevenindo o surgimento de infecçoes de repetiçao. O tratamento com rHUG-CSF aumenta o número de granulócitos, diminui o número de novas infecçoes e melhora de forma significativa a sobrevida e qualidade de vida. O transplante de células-tronco hematopoiéticas é indicado em casos refratários ao tratamento com rHUG-CSF que apresentam infecçoes recorrentes graves e resistência ao tratamento sem detecçao de mielodisplasia/leucemia.
Descritores: Neutropenia, neutropenia congênita, ELANE, G6PC3, síndrome de Shwachman-Diamond.
INTRODUÇAO
Os neutrófilos, ou leucócitos polimorfonucleares neutrofílicos sao a populaçao mais abundante de leucócitos no sangue que medeiam as fases iniciais da resposta inflamatória. Durante o desenvolvimento fetal, os leucócitos sao produzidos primariamente no fígado fetal nos dois primeiros meses de vida, e após o quinto mês de gestaçao, esta funçao passa a ser realizada na medula óssea. Os neutrófilos sao produzidos a partir de células precurssoras da medula óssea, comprometidas para a linhagem mieloide1. A mielopoiese é um processo que ocorre no microambiente estromal da medula óssea, auxiliado por citocinas como o fator de crescimento de granulócitos (G-CSF), fator de crescimento de monócitosgranulócitos (GM-CSF), fator de células precursoras(SCF), interleucina-3(IL-3) einterleucina-6(IL-6). Sao produzidos por linfócitos T, monócitos, fibroblastos, células endoteliais e reticuloendoteliais. Os estágios de maturaçao dos neutrófilos sao mieloblasto, promielócito, neutrófilo mielócito, neutrófilo metamielócito, neutrófilo bastonete e finalmente neutrófilo segmentado2. Durante os estágios de diferenciaçao mieloide dos neutrófilos, ocorre perda do receptor CD34 (marcador de células-tronco), com transitória expressao de HLA-DR, surgimento de CD13 e CD38 nos mieloblastos. No estágio de mieloblasto e promielócito nao ocorre expresssao de CR3 e FcgRIII, que estao presentes nos estágios subsequentes da mielopoiese (Tabela 1). Sob condiçoes fisiológicas, os neutrófilos necessitam de 9 a 11 dias para sofrerem maturaçao e atingirem a circulaçao sanguínea. Os estágios de mieloblastos e promieloblastos levam 18 a 24 horas para se desenvolverem, o mielócito 52 horas; e para alcançarem a circulaçao sanguínea, 96 a 144 horas. Em condiçoes basais, 6,0 x 107 neutrófilos/kilograma de peso vivo sao repostos a cada hora. Em situaçoes de stress e infecçao o turnover é acelerado1,2. A maturaçao dos neutrófilos na medula óssea ocorre em duas situaçoes distintas; ontogenia fisiológica e resposta a infecçoes. Morfologicamente, os neutrófilos apresentam aparência esférica com 12 a 15 µm de diâmetro, núcleo segmentado com três a cinco lóbulos conectados e citoplasma rico em três tipos de grânulos, específicos, azurófilos e terciários. Os grânulos primários ou azurófilos sao expressos a partir do estágio promielócito, sendo ricos em hidrolases, proteases e mieloperoxidase. Os grânulos secundários ou específicos sao expressos no estágio metamielócito, e contém lisozima, colagenase e elastase. Os grânulos terciários contêm gelatinases, catepsinas e glicoproteínas, sendo considerados por alguns autores variaçao morfológica dosgrânulos primários ou secundários, ou artefatospor contaminaçao de células polimorfonucleares neutrofílicas por monócitos1(Tabela 2). Depois de maturos, os neutrófilos circulam em média 6 a 10 horas no sangue e sobrevivem por até 48 horas nos tecidos inflamados. Neutrófilos circulantes correspondem a apenas 3 a 5% de todos os neutrófilos no corpo humano, cujo número total pode chegar a 3,5 x 108 células/kilograma de peso vivo. Se nao forem recrutados para um local inflamado dentro desse período, eles sofrem apoptose e fagocitose por macrófagos residente do fígado ou baço. Durante a resposta inflamatória, os neutrófilos se acumulam ao longo da superfície endotelial, migram entre as junçoes intercelulares da regiao pós-capilar e atingem o tecido inflamado, onde causam a morte e destruiçao dos patógenos3.
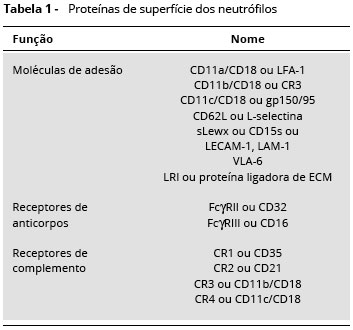
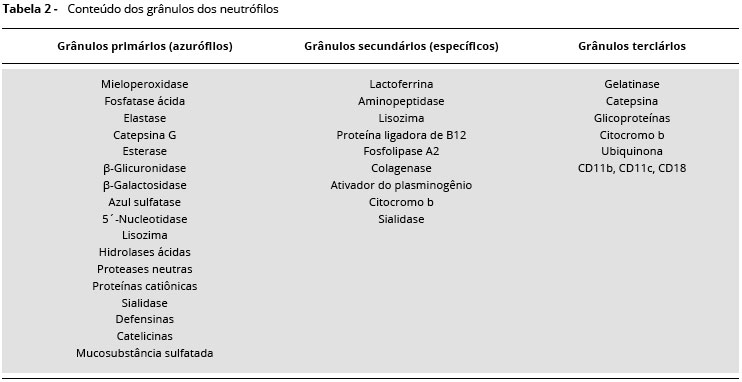
NEUTROPENIA
O número de neutrófilos circulantes no recémnascido é elevado nas primeiras 72 horas de vida e perdura durante os dois primeiros meses, com gradual decréscimo a partir dessa idade. A contagem de neutrófilos em neonatos é extremamente variável (12.000 a 15.000 neutrófilos/mm3de sangue)4. Partos com mais de 12 horas de duraçao estao associados a alto número de neutrófilos no sangue, ao passo que partos prematuros, com menos de 32 semanas de gestaçao, estao associados com baixo número de neutrófilos no sangue. O número de neutrófilos pode variar em situaçoes de stress, sazonalidade ou patologias2. Neutropenia é definida como reduçao do número absoluto de neutrófilos na circulaçao sanguínea (abaixo de 2.000 neutrófilos/mm3 de sangue em crianças entre 2 a 12 meses de idade e abaixo de 1.500 neutrófilos/mm3 de sangue em crianças com mais de um ano de idade),1,4 sendo considerada grave quando a contagem for inferior a 500 neutrófilos/mm3 de sangue, e crônica quando permanece baixa durante os últimos 3 meses.
A neutropenia pode ser classificada como de grau leve (1.000-1.500 neutrófilos/mm3 de sangue), moderado (500-1.000 neutrófilos/mm3 de sangue) ou grave (< 500 neutrófilos/mm3 de sangue)1. A neutropenia é confirmada através da realizaçao de leucogramas repetidos, sendo necessários três por semana, durante seis semanas para a neutropenia cíclica. A neutropenia é considerada permanente se observada em todas as amostras, ou intermitente se sao observados períodos de normalizaçao espontânea; e cíclica se ocorre a intervalos periódicos de 21 dias (podendo variar de 14-36 dias)5. A neutropenia é considerada central quando existe depleçao celular da medula óssea, com deficiência dos estágios iniciais de maturaçao, ou periférica se a maturaçao dos neutrófilos na medula óssea é normal6.
Monocitose, eosinofila e hipergamaglobulinemia podem estar associadas com a neutropenia, de maneira inversa com a gravidade da doença. A monocitose compensatória está associada com boa tolerância clínica a infecçoes em inúmeras formas de neutropenia5. A frequência e a gravidade das infecçoes dependem nao só da contagem e velocidade de queda do número de neutrófilos, mas também de anormalidades da funçao fagocitária, déficits da funçao imune adaptativa, condiçoes do hospedeiro e germe específico. Pacientes com neutropenia crônica grave apresentam alto risco de infecçao, da mesma maneira que pacientes com neutropenia por depleçao celular da medula óssea. Na neutropenia central, o risco de infeçao é baixo em contagens acima de 1.000 neutrófilos/mm3 de sangue, moderado entre 1.000 a500 neutrófilos/mm3 de sangue e alto em contagens abaixo de 500 neutrófilos/mm3 de sangue4. Os locais mais afetados sao pele e mucosas e do trato digestório, respiratório e genitourinário. Distúrbios orais sao frequentes em pacientes com neutropenia central e crônica grave, na forma de estomatite erosiva e hemorrágica, gengivite e úlceras na língua. Lesoes gastrintestinais sao comuns, causando dor abdominal e diarreia, mimetizando Doença de Crohn, e estando associadas àinfecçaoe ulceraçao perianal. Os principais agentes sao o Staphylococcus aureus, S. epidermitis, Streptococcus spp., Enterococcus spp., Burkholderia cepacia, Nocardia asteroides, Pneumococcus spp., Pseudomonas aeruginosa, bacilos gram-negativos, Candida albicans e Aspergilluss spp.1.
CLASSIFICAÇAO DAS NEUTROPENIAS CONGENITAS
Nao existe um consenso absoluto para a classificaçao das neutropenias congênitas, sendo a genotipagem a informaçao mais importante para distinguir a neutropenia congênita de outras formas de neutropenia. O termo neutropenia congênita nao é utilizado de forma homogênea6, nao sendo utilizado quando acompanhado de alteraçoes da imunidade adaptativa ou extra-hematopoiéticas. Na Tabela 3 estao descritas neutropenias congênitas segundo a presença ou ausência de manifestaçoes extra-hematopoiéticas e defeitos associados.
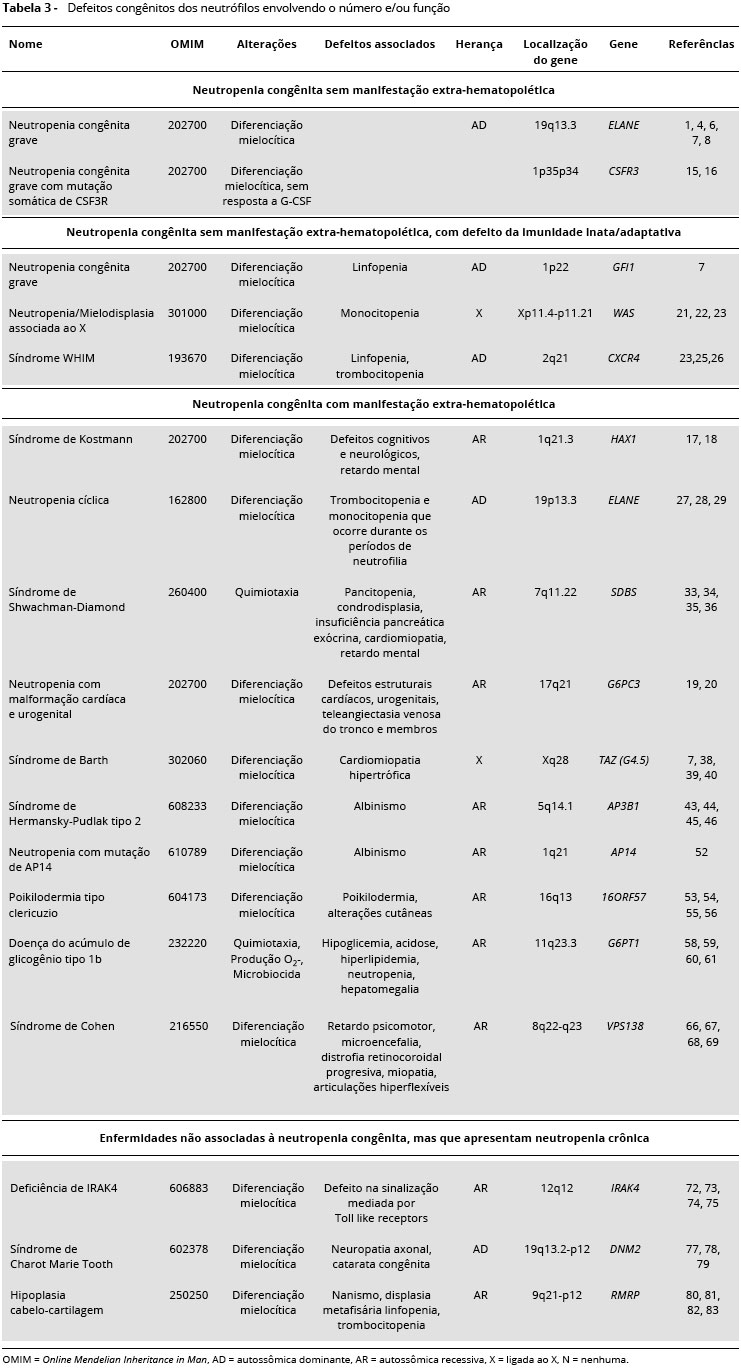
Neutropenia congênita grave
A neutropenia congênita grave corresponde a um grupo genético heterogêneo1,4,6 que afeta a diferenciaçao mieloide de neutrófilos, acometendo 3-4/1x106 indivíduos. A forma autossômica dominante pode ser causada por mutaçao do gene ELANE, localizado no cromossomo 19q13.3, ou por mutaçao do gene CSF3R localizado no cromossomo 1p35p34, ambas consideradas neutropenia congênita sem manifestaçao extra-hematopoiética. Outras formas de neutropenia congênita grave incluem a mutaçao do gene GFI1 localizado no cromossomo 1p22, na forma de herança autossômica dominante; e mutaçao do gene HAX1 localizado no cromossomo 1q21.3na forma autossômica recessiva. Outra forma de herança autossômica recessiva é causada por mutaçao do gene G6PC3 localizado no cromossomo 17q21; e a mutaçao do gene WAS localizado no cromossomo Xp116 é ligada ao cromossomo X. A neutropenia congênita grave por mutaçao de GFI1 e WAS é acompanhada de defeito da imunidade inata ou adaptativa sem alteraçoes extra-hematopoiéticas, ao passo que a mutaçao G6PC3 causa neutropenia congênita com manifestaçao extrahematopoiética. Pacientes com neutropenia congênita grave apresentam contagem abaixo de 500 neutrófilos/mm3 de sangue, febre, infecçoes recorrentes do trato respiratório, digestório e pele no primeiro ano de vida7. Vinte por cento dos pacientes desenvolvem leucemia ou síndrome mielodisplásica durante a adolescência8.
Neutropenia congênita grave com mutaçao de ELANE
Esta enfermidade é causada por mutaçoes do gene ELANE9 localizado no cromossomo 19p13.3110. O gene ELANE codifica a proteína elastase 2 de neutrófilos, uma serino protease que hidrolisa a elastase e a enzima α1-antitripsina, possuindo homologia com outras duas proteases produzidas por células polimorfonucleares: proteinase 3 e azurocidina.Estas trêsproteínas,elastase 2, proteinase 3 e azurocidina sao codificadas por genes localizados no cromossomo 19p13.3 e sao reguladas conjuntamente, o que explica seu envolvimento na neutropenia congênita grave e neutropenia cíclica. A elastase 2 está presente no interior dos grânulos específicos ou azurófilos, e degrada a proteína da membrana externa A (OmpA) da Escherichia coli e fatores de virulência das bactérias Shigella, Salmonella e Yersinia. Mutaçoes em ELANE causam apoptose prematura de mielócitos, interrompendo o ciclo normal de maturaçao11. A mutaçao ELANE é observada em 50% dos pacientes com neutropenia congênita grave, e em pacientes com neutropenia cíclica. Na neutropenia congênita grave sao observadas infecçoes bacterianas e fúngicas recorrentes, estomatite, neutropenia com menos de 500 neutrófilos/mm3 de sangue, monocitose, eosinofilia, hipergamaglobulinemia e distúrbios na maturaçao dos neutrófilos na medula óssea12. O tratamento destes pacientes consiste na utilizaçao do fator estimulador de colônia de granulócitos recombinante (rHUG-CSF), que aumenta o número de granulócitos circulantes no sangue, diminui o número de infecçoes e melhora de forma significativa a sobrevida e qualidade de vida6. A literatura descreve o surgimento de mielodisplasia13 ou leucemia mieloide aguda 14 em uma frequência muito baixa após tratamento prolongado com rHUG-CSF em pacientes com mutaçao em ELANE. Embora esta associaçao nao se encontre bem estabelecida, pacientes com mutaçao em ELANE sob tratamento prolongado com rHUG-CSF devem ser monitorados para o desenvolvimento de leucemia mieloide aguda14.
Neutropenia congênita grave com mutaçao somática de CSF3R
Esta é uma neutropenia congênita cujo fenótipo clínico é semelhante à neutropenia congênita grave por mutaçao em ELANE. Pacientes com mutaçao em CSFR3 apresentam alto risco de evoluçao para síndrome mielodisplásica e/ou leucemia mieloide aguda15 e sao incapazes de responder à terapia com rHUG-CSF em doses superiores a 100 µg/kg/dia. Camundongos knock-in para CSF3R16 reproduzem a mutaçao observada em seres humanos, e o tratamento com rHUG-CSF aumenta o número de neutrófilos circulantes sem risco de desenvolvimento de leucemia mieloide aguda ou mielodisplasia nestes animais13.
Neutropenia congênita grave com mutaçao de GFI1
Esta é uma neutropenia congênita grave com herança autossômica dominante por mutaçao do gene GFI1, localizado no cromossomo 1q22, que codifica um fator de transcriçao repressor da enzima elastase, que afeta a diferenciaçao mieloide. Além de agranulocitose, é observado neste grupo de pacientes interrupçao da maturaçao na fase promielocítica, linfopenia de células T e B, hipogamaglobulinemia, anemia e trombocitopenia. Pacientes com mutaçao em GFI1 apresentam infecçoes bacterianas recorrentes a partir dos 3 meses de idade, principalmente na boca e regiao perineal7.
Síndrome de Kostmann
A síndrome de Kostmann, neutropenia congênita grave autossômica recessiva tipo 3 ou agranulocitose genética infantil, é uma herança autossômica recessiva commutaçao no gene HAX117,localizado no cromossomo 1q21.3. Este gene desempenha um papel significativo na apoptose de neutrófilos, impedindo a diferenciaçao dos neutrófilos nos estágios promielócito e mielócito. A enfermidade é caracterizada por início precoce de neutropenia extremamente grave (< 200 neutrófilos/mm3 de sangue), infecçoes bacterianas graves e morte de crianças com menos de 3 anos de idade18. As infecçoes bacterianas comumente envolvem seios nasais, pulmao, fígado, pele e mucosa oral. Aproximadamente 40% dos pacientes apresentam diminuiçao da densidade óssea e osteoporose. Uma pequena porcentagem de pacientes apresenta defeito cognitivo e neurológico associado à neutropenia17. O tratamento destes pacientes consiste no uso de rHUG-CSF, permitindo que a contagem de neutrófilos atinja 1.000 neutrófilos/mm3 de sangue. Em casos graves e refratários é indicado o transplante de medula óssea.
Neutropenia com malformaçao cardíaca e urogenital
A neutropenia com malformaçao cardíaca e urogenital é uma enfermidade autossômica recessiva causada por mutaçao do gene G6PC319, localizado no cromossomo 17q21, que inibe a atividade da enzima glicose-6-fosfatase e aumenta a apoptose de neutrófilos e fibroblastos. Crianças com neutropenia com malformaçao cardíaca e urogenital apresentam início precoce de infecçoes bacterianas recorrentes graves e neutropenia extremamente grave (< 200 neutrófilos/mm3 de sangue), onde 75% das crianças afetadas morrem antes dos 3anos de idade. Os pacientes afetados apresentam defeitos cardíacos ou malformaçao urogenital, veias subcutâneas proeminentes e angiectasia19,20. O tratamento consiste na utilizaçao de rHUG-CSF.
Neutropenia/Mielodisplasia ligada ao X
A neutropenia/mielodisplasia ligada ao X é uma neutropenia congênita com padrao de herança ligada ao cromossomo X, causada por mutaçao no gene WAS21, localizado no cromossomo Xp11.4-p11.2122. O WAS é o gene responsável pela regulaçao da mobilizaçao da actina do citoesqueleto dos leucócitos, causando interrupçao da maturaçao no estágio promielócito/mielócito23. Os pacientes apresentam infecçoes bacterianas recorrentes graves nos primeiros meses de vida, neutropenia grave (< 500 neutrófilos/mm3 de sangue), monocitose ou monocitopenia, diminuiçao do número de células NK CD3-CD16+CD56+, linfopenia de células B, aumento do número de células TCD8, e desequilíbrio da proporçao CD4/CD8 para abaixo de 0,521, a despeito da quantidade normal de linfócitos T CD4. A literatura reporta casos de baixos níveis séricos de IgA21 e trombocitopenia23. Uma vez que estes pacientes apresentam displasia de células da medula óssea (megacariócitos gigantes, micromegacariócitos, hipogranularidade, reduçao da granulopoiese e número excessivo de blastos), podem desenvolver leucemia22. O tratamento envolve o uso profilático de antibiótico, e o rHUG-CSF é recomendado em casos de neutropenia grave com risco de sepse.
Síndrome WHIM
A síndrome WHIM é causada por mutaçao do gene CXCR4 localizado no cromossomo 2q22.123 que codifica o receptor CXCR4 para a quimiocina CXCL12. É uma herança autossômica dominante cujo gene afetado está envolvido no processo de organogênese, ontogenia dos linfócitos B e mielopoiese. Sao comuns nestes pacientes neutropenia, hipogamaglobulinemia, infecçao recorrente do trato respiratório e displasia cervical e vulvar25. Esta enfermidade foi descrita inicialmente em uma família cujo pai e duas filhas apresentavam infecçao crônica por papilomavírus, infecçao bacteriana sinopulmonar, linfopenia, hipogamaglobulinemia, hipercelularidade da medula óssea e neutropenia com anormalidade morfológica (núcleo hipersegmentado, citoplasma vacuolizado). A ocorrência de neutropenia e retençao de neutrófilos na medula óssea é denominada myelokathexis (kathexis = retençao)26.
Neutropenia cíclica
A neutropenia cíclica é uma enfermidade autossômica dominante27 causada por mutaçao do gene ELANE, localizado no cromossomo 19p13.328 acometendo 1/1x106 indivíduos. A enfermidade é caracteriza por períodos de neutropenia (< 1.000 neutrófilos/mm3 de sangue) com duraçao de 3-10 dias que se repetem em intervalos de 21 dias29, podendo variar de 14-36 dias. A neutropenia é acompanhada por trombocitopenia, eosinofilia, linfocitose e monocitose5. Os sintomas mais comuns durante a neutropenia sao febre, mal-estar, periodontite, ulceraçao da mucosa oral, impetigo e linfadenopatia, sintomas comuns em crianças e adolescentes, ao passo que adultos apresentam neutropenia leve a moderada sem ciclos bem definidos. Celulite, enterocolite necrosante e bacteremia sao complicaçoes graves que podem ser fatais27. O diagnóstico é realizado através de hemogramas, duas vezes por semana durante 6 semanas. A maioria dos pacientes com neutropenia cíclica durante os surtos de neutropenia e infecçoes associadas sao tratados com antibióticos ou imunoglobulinas para uso endovenoso (IgEV). Em casos graves, o tratamento consiste na utilizaçao derHUG-CSF em pacientes sintomáticos27, aumentando a contagem de neutrófilos para mais de 1.000 neutrófilos/mm3 de sangue. Embora a maioria dos pacientes nao necessitem de rHUG-CSF, nos casos graves de neutropenia cíclica nao responsivos a terapia com rHUG-CSF é indicado o transplante de medulaóssea30. Modelo animal de estudo da enfermidade é encontrado em caes da raça Collie cinza como herança autossômica recessiva31 causada por mutaçao no gene que codifica a subunidade beta da proteína adaptadora do complexo-3 (AP3), responsável pela exportaçao de proteínas do trans-Golgi para os lisossomos32.
Síndrome de Shwachman-Diamond
A síndrome de Shwachman-Diamond é uma enfermidade autossômica recessiva com mutaçao do gene SBDS33 localizado no cromossomo 7q11.2234, caracterizada por pancitopenia, insuficiência pancreática exócrina, condrodisplasia, susceptibilidade a infecçoes, leucemia e anormalidades esqueléticas. Sua incidência tem sido estimada em 1/75.000 indivíduos. O gene SBDS é responsável pela sobrevivência dos precursores granulocíticos e quimiotaxia de neutrófilos. Esta síndrome é a segunda causa mais comum de insuficiência pancreática após a fibrose cística e a terceira causa hereditária de insuficiência da medula óssea após a anemiade Fanconi e anemia de Blackfan-Diamond. Os pacientes apresentam na primeira infância má absorçao, esteatorreia e retardo do crescimento. Neutropenia é a anormalidade hematológica mais comum, observada em 98% dos pacientes, seguido de anemia (42%), trombocitopenia (34%) e pancitopenia (19%). Infecçoes bacterianas do trato respiratório, otite média, sinusite, pneumonia, estomatite, paroníquia, osteomielite e bacteremia sao comuns35. Anomalias esqueléticas sao relatadas em mais de 75% dos pacientes, e mais de 50% destes indivíduos têm baixa estatura com velocidade de crescimento normal. Distúrbios cognitivos e graus variáveis do desenvolvimento mental sao observados em 15% dos pacientes36. A morte geralmente ocorre por sepse ou neoplasia. O tratamento envolve o uso de antibiótico, e o uso de rHUG-CSF em pacientes com a síndrome de Shwachman-Diamond é menos frequente. Estes pacientes necessitam de atençao especial, uma vez que a insuficiência pancreática pode levar ao surgimento de déficit nutricional e de vitaminas liposolúveis6.
Síndrome de Barth
A síndrome de Barth, ou miopatia cardioesquelética com neutropenia e anormalidade mitocondrial ou acidúria 3-metilglucataconica tipo II é uma enfermidade ligada ao cromossomo X com mutaçao no gene TAZ, localizado no cromossomo Xq28. Esta enfermidade foi descrita inicialmente por Barth et al.37 como uma herança ligada ao cromossomo X cujos pacientes apresentavam cardiomiopatia dilatada, fibrose endomiocardial, miopatia, neutropenia e anormalidade mitocondrial (mitocôndrias concêntricas, com corpos de inclusao e cristas alongadas)38. Também apresentam acidose metabólica por alteraçao do metabolismo do ácido 3-metilglutaconico e 2-etilidracrilico39. A apresentaçao inicial da doença varia desde uma cardiomiopatia dilatada congênita na infância à insuficiência cardíaca congestiva, infecçoes bacterianas recorrentes e neutropenia40. A síndromede Barth é a primeira doença descrita com erro inato do metabolismo afetando a cardiolipina, um componente da membrana interna das mitocôndrias necessário para o transporte na cadeia de elétrons41. Os pacientes possuem diminuiçao da cardiolipina total e subclasses, como cardiolipina-tetralineoyl. Para o tratamento dos sintomas cardíacos, os digitálicos nao sao efetivos e a suplementaçao com L-carnitina causa rápida deterioraçao do status cardíaco. Acido pantotênico, um precursor da coenzima A melhora a funçao cardíaca, contagem de neutrófilos, hipocolesteloremia e hiperucemia6. O diagnóstico da síndrome de Barth é baseado na tríade cardiomiopatia dilatada, neutropenia e aumento dos níveis do ácido 3-metilglutacônico37-39. Um modelo desta síndrome foi desenvolvido na Drosophila melanogaster, acarretando reduçao da atividade locomotora e anormalidades das cristas mitocondriais42.
Síndrome de Hermansky-Pudlak tipo 2
A síndrome de Hermansky-Pudlak tipo 2 (HPS2)43 é uma enfermidade autossômica recessiva com mutaçao no gene AP3B144, localizado no cromossomo 5q14.145 responsável pelo tráfico de proteínas do trans-Golgi e compartimento tubular endossomalao compartimento endossomo/lisossomo. Esta mutaçao causa perda da subunidade 3A deste complexo transportador46 e aumento da expressao de CD63 na membrana dos lisossomos47. Os pacientes apresentam albinismo oculocutâneo parcial, complicaçoes hemorrágicas, plaquetas granuladas, neutropenia48, leve retardo mental6, periodontite e queda de dentes45. Pacientes com HPS2 apresentam defeito na ligaçao de CD1B ao complexo proteico adaptador AP3, causando defeito na apresentaçao de antígenos lipídicos49, perda de microtúbulos responsáveis pela movimentaçao de perforinas e granzimas até a sinapse inumológica e defeito na atividade citotóxica de células T CD850 e NK51. A HPS2 difere das outras formas de HPS pelo aumento na susceptibilidade a infecçoes45. O tratamento com rHUG-CSF previne o surgimento de infecçoes bacterianas, embora tenha sido descrito caso de linfohistiocitose hemofagocítica fulminante e resistência ao tratamento com rHUG-CSF48.
Neutropenia com mutaçao de AP14
Esta enfermidade autossômica recessiva é causada por mutaçao do gene AP14, localizado no cromossomo 1q21, responsável pelo empacotamento lisossomal. A proteína adaptadora p14 está associada com a proteína quinase ativada por mitógeno (MAPK) que sinaliza para o endossomo tardio, importante para a funçao de neutrófilos, células B, TCD8 e melanócitos. Os pacientes acometidos apresentam albinismo parcial, neutropenia grave e susceptibilidade a infecçao por pneumococos52.
Poikilodermia de Clericuzio com neutropenia
A Poikilodermia de Clericuzio com neutropenia53 é uma enfermidade autossômica recessiva que afeta o gene 16ORF5754 localizado no cromossomo 16q13, apresentando várias similaridades com a síndrome Rothmund-Thompson, como manifestaçoes esqueléticas, catarata e predisposiçao a osteossarcoma55,56. Os pacientes apresentam atrofia cutânea, eritema papular e genodermatose. As lesoes aparecem no início da vida na forma de rash que gradualmente se propagam para os membros e se transformam em placas hipo ou hiperpigmentadas comteleangiectasia54. Ocorre neutropenia grave com interrupçao da maturaçao dos neutrófilos no estágio promielócito, neutropenia cíclica ou de apresentaçao variável. Pode ocorrer hipergamaglobulinemia policlonal e infecçao broncopulmonar55.
Doença do acúmulo do glicogênio tipo 1b
A doença do acúmulo do glicogênio tipo 1b58 é uma doença autossômica recessiva causada por mutaçao no gene G6PT159, localizado no cromossomo 11q23.360, que causa deficiência na produçao do transportador 1 de glicose-6-fosfato, responsável pelo transporte da glicose6-fosfato para o lúmen do retículo endoplasmático. No retículo endoplasmático está localizada a unidade catalítica da G6Pase, responsável pela manutençao da glicemia sanguínea. A atividade da G6Pase necessita de dois componentes da membrana microssomal: um transportador específico do sistema glicose-6-fosfato que lança G6P do citoplasma para o lúmen do retículo endoplasmático (uma G6P translocase), e a enzima glicose-6-fosfato fosfohidrolase, ligada à superfície luminal da membrana. Os pacientes apresentam hepatoesplenomegalia, hipoglicemia, acidose láctica com atividade latente normal de glicose-6-fosfato no fígado, hiperlipidemia, doença inflamatória intestinal, estomatite, abscessos e neutropenia61. Defeitos na motilidade, quimiotaxia, burst oxidativo e morte microbiana sao atribuídos ao deficiente metabolismo da via hexose monofosfato e glicólise anaeróbica62,63. Uma vez que os neutrófilos sao responsáveis pela defesa de barreiras naturais como as mucosas, infecçao e ulceraçao oral e anal, pneumonia e septicemia sao comuns64. Os principais agentes sao Staphylococcus aureus, Streptococcus do grupo A e Escherichia coli. O uso de rHUG-CSF na dose de 5 µg/kg/dia é indicado para a correçao da neutropenia e controle de infecçoes graves, sendo obtidos bons resultados após 48 horas de tratamento65. A literatura reporta alguns pacientes cujo uso prolongado de rHUG-CSF esteve associado a trombocitopenia e esplenomegalia.
Síndrome de Cohen
Esta síndrome autossômica recessiva com mutaçao do gene VPS13B66 localizado no cromossomo 8q22-q2367 está associada com moderado a grave retardo psicomotor, microencefalia, anormalidade facial, miopatia, hipotonia, distrofia retinocoroidal progressiva68, obesidade do tronco, hiperextensao de ligamentos, prolapso de valva mitral, refluxo gastroesofágico e hérnia de hiato69. A neutropenia está presente em mais de 90% dos casos relatados, sendo responsável pelo desenvolvimento de infecçoes crônicas da pele e gengivoestomatite67. O diagnóstico diferencial inclui síndrome de Marfan, síndrome de Sotos, hipotireoidismo, e retardo mental sem causa estabelecida. Pode ocorrer trombocitopenia transitória ou persistente70. Para o correto diagnóstico é fundamental a presença de retardo psicomotor nao progressivo, desarranjo motor, microencefalia, aspecto facial típico, hipotonia infantil, hiperextensao das articulaçoes, alteraçoes oculares (distrofia retinocoroidal em pacientes com mais de 5 anos de idade) e períodos isolados de granulocitopenia68,70. O fenótipo muda com o passar da idade, e as alteraçoes psicomotoras sao profundas em 22%, moderada em 6%, e leve em 11% dos pacientes68,71.
Deficiência de IRAK4
A deficiência de IRAK4 é uma enfermidade autossômica recessiva causada pela mutaçao do gene IRAK4 localizado no cromossomo 12q12, que codifica o receptor de interleucina-1 associado à kinase 4 (IRAK4), que causa defeito na sinalizaçao mediada por TLR4/IL1R72. Os pacientes apresentam infecçoes piogênicas recorrentes, celulite, osteomielite e baixa resposta inflamatória73. Os agentes mais comuns sao Streptococcus pneumoniae e Staphylococcus aureus. A maioria dos pacientes apresenta níveis de anticorpos contra proteínas e polissacarídeos normais, embora tenham sido descritos pacientes com baixa resposta ao LPS ou S. aureus74. PBMC destes pacientes respondem normalmente ao TNF-α, mas nao a IL-1ß, IL-18, e agonistas de TLR1-6 ou TLR9. Pacientes com mutaçao em IRAK4 apresentam neutropenia moderada que tende a se normalizar durante os períodos de infecçao75, e seus neutrófilos produzem baixas quantidades de ânion superóxido76. Alguns pacientes sao incapazes de sustentar uma resposta mediada por anticorpos contra polissacarídeos e proteinas, apresentando infecçoes bacterianas e fúngicas recorrentes, necessitando muitas vezes de terapia intravenosa com imunoglobulinas73,74.
Síndrome de Charot-Marie Tooth
A síndrome de Charcot-Marie-Tooth (CMT) é uma enfermidade autossômica dominante com mutaçao no gene DNM277 localizado no cromossomo 19p13.278. Esta síndrome apresenta grande heterogeneidade clínica e genética, afetando o sistema nervoso periférico, causando fraqueza progressiva e atrofia muscular. A CMT neuropática é subdividida em CMT1 e CMT2 baseado em critérios clínicos e eletrofisiológicos. A CMT1 ou neuropatia sensorial ou motor hereditária do tipo I (HMSN I) é uma neuropatia desmielinizante, ao passo que a CMT2 ou HMSN II é uma neuropatia axonal79. Outras formas dominantes da doença incluem CMTDIC, causada por mutaçao do gene YARS localizado no cromossomo 1p35-p34.1; CMTDID, causado por mutaçao do gene MPZ localizado no cromossomo 1q22; e CMTDIE causado por mutaçao do gene INF2 localizado no cromossomo 14q. A neutropenia nestes pacientes é de moderada e levemente sintomática, embora possa ser grave no início da infância78.
Hipoplasia cabelo-cartilagem
A hipoplasia cabelo-cartilagem80 é uma enfermidade autossômica recessiva que afeta o gene RMRP81 localizado no cromossomo 9q21-p1282, com incidência estimada em 1/23.000 nascidos vivos. As alteraçoes esqueléticas nos incluem nanismo, membros curtos, maos pequenas, hiperextensibilidade articular (especialmente maos, punhos e pés), extensao limitada dos cotovelos, genu varum, encurtamento do fêmur, tíbia menor que a fíbula, displasia metafisária e deformidade torácica. A histopatologia óssea demonstra hipoplasia cartilaginosa. Outros sinais clínicos incluem cabelos finos e esparsos, braquicefalia ocasional, lordose lombar, escoliose leve, atresia do esôfago e doença de Hirschsprung83. Os pacientes apresentam susceptibilidade a varicela, herpes simplex, linfopenia de células T CD4 e NK84, baixa resposta proliferativa a mitógenos policlonais como fitohemaglutinina (PHA), cocanavalina A (Con-A), pokeweed mitogen (PWM)85. Sao comuns anemia macrocítica86 e trombocitopenia87. Também é observada má absorçao intestinal e megacólon88. Pode ocorrer linfoma, carcinoma basocelular, carcinoma hepático primário e carcinoma duodenal89,90. Pneumonia por Streptococcus pneumoniae, Klebsiella pneumoniae89 e bronquiectasia84 sao comuns. O tratamento pode incluir o uso de interferon recombinante humano (rHU-IFN) em crianças com varicelaou imunossuprimidos com câncer91,ou aciclovir para varicela92, sendo contraindicado o uso de vacina contra poliomielite contendo vírus vivo atenuado.
Outras enfermidades que podem apresentar neutropenia
Neutropenia também pode ser observada em pacientes com síndrome de Bruton93, deficiência de CD40L94, síndrome do linfócito desnudo tipo II 95, ataxia-teleangiectasia96,síndrome de Wiskott-Aldrich97, disgenesia reticular98, síndrome 22q1199, síndrome de Chédiak Higashi94, síndrome de Grisceli tipo 2 (GS2)100, linfohistiocitose fagocítica familiar101, anemia de Blackfan-Diamond102, anemia de Fanconi103, Propionyl-CoA Carboxylase deficiência104, síndrome de Pearson105, febre familiar do Mediterrâneo106 e síndrome nefrótica do tipo Finlandês 1 (NPHS1)107 (Tabela 4).
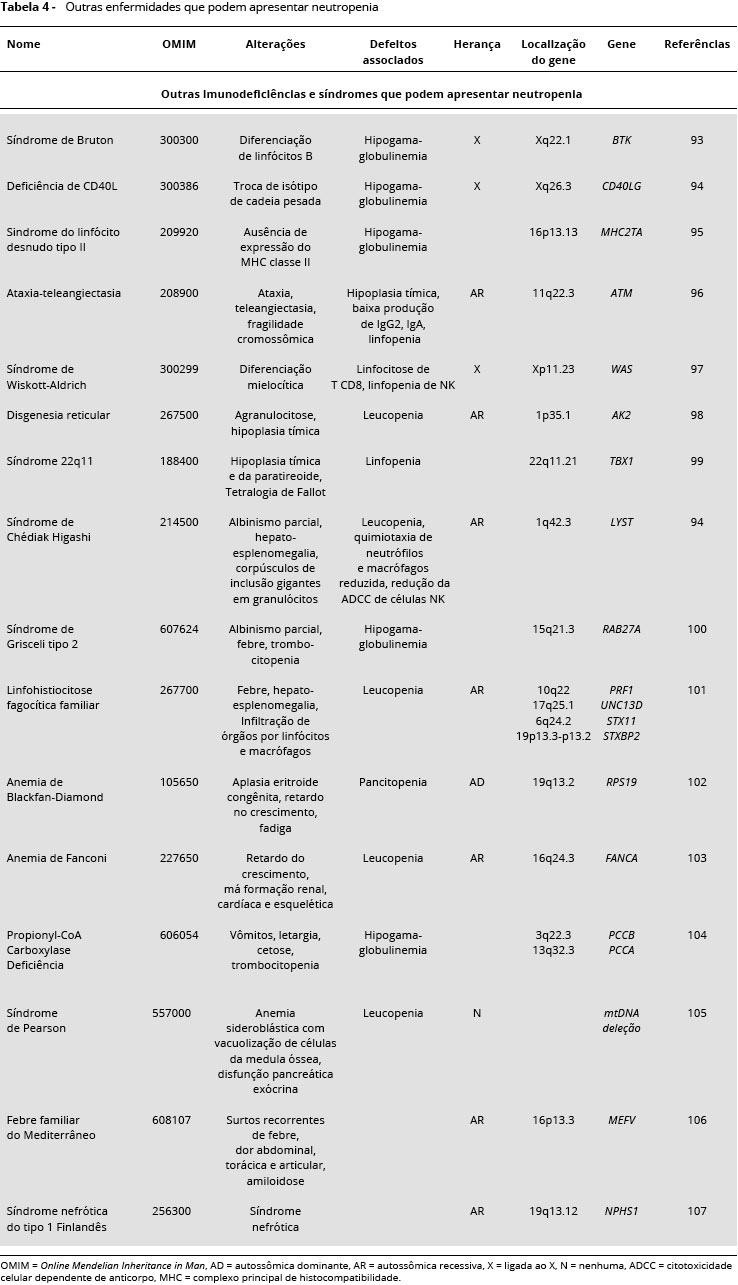
DIAGNOSTICO DA NEUTROPENIA CONGENITA
O diagnóstico de neutropenia congênita é suspeito em crianças com infecçao bucal recorrente e grave associada com neutropenia e infecçao bacteriana ou fúngica persistente. O mielograma é fundamental para o diagnóstico, na busca de neoplasia hematológica, maturaçao mieloide, hipereosinofilia, monocitose, hemofagocitose e myelokathexis. Outros exames incluem níveis de anticorpos antineutrófilos, dosagem de imunoglobulinas séricas, imunofenotipagem, marcadores pancreáticos (tripsinogênio sérico, elastase fecal) e dosagem de vitaminas liposolúveis (vitamina A, E, D)6. Como diagnóstico diferencial, deve-se investigar neutropenia aloimune, muitas vezes observada durante o nascimento, sendo inicialmente grave (< 100 neutrófilos/mm3 de sangue), cuja contagem normaliza após 3 a 6 meses de idade, causada por incompatibilidade materno-fetal pela sensibilizaçao da mae com antíge nos derivados de neutrófilos. Outra causa comum de neutropenia em crianças é a neutropenia autoimune primitiva ou neutropenia crônica benigna, cujos pacientes apresentam quadro moderado a grave de infecçao com menos de 8 meses de idade, acompanhada de monocitose, eosinofilia e esplenomegalia. Esta neutropenia pode desaparecer espontaneamente após 12 a 36 meses. Outras causas de neutropenia incluem lúpus eritematoso sistêmico, artrite reumatoide, síndrome de Evans e neutropenia idiopática. Estudos iniciados na década de 40 demonstraram que grupos étnicos nao brancos (africanos, afro-caribenhos, afro-americanos e seus descendentes) apresentam como característica racial contagem de leucócitos e neutrófilos mais baixa, sendo esta variaçao denominada neutropenia étnica benigna, pseudoneutropenia, neutropenia benigna dos negros, neutropenia hereditária benigna e leucopenia e neutropenia familiar benigna. Tal condiçao pode ser interpretada muitas vezes de forma errônea, levando a investigaçoes desnecessárias. A neutropenia étnica benigna é frequente em pessoas da raça negra (4,5%), seguida de caucasianos (0,8%), apresentando contagem entre 500 a 1.500 neutrófilos/mm3 de sangue, ausência de infecçoes ou sintomas clínicos1,4,6,7.
PROFILAXIA E TRATAMENTO
Pacientes com neutropenia congênita nao sao susceptíveis a infecçoes virais. Nestes pacientes podem ser utilizadas vacinas contendo vírus vivos atenuados, sendo recomendado o uso de vacinas contra influenza e até pneumococos, mas contraindicado o uso de BCG6. A profilaxia consiste no uso de antibióticos de amplo espectro, principalmente sulfametazol/trimetropina 50mg/kg/dia, segundo experiência como tratamento de pacientes com Doença Granulomatosa Crônica(DGC). O uso de antibióticos endovenosos e profiláticos aumentas significativamente o tempo de sobrevida dos pacientes, embora a qualidade seja baixa, em funçao do número de infecçoes recorrentes4. O uso de concentrado de granulócitos é restrito a casos de celulite ou infecçoes bacterinas e fúngicas que nao respondem à antibióticoterapia. De maneira geral, em pacientes com DGC, nao é recomendável a transfusao de concentrado de granulócitos profilática, uma vez que a meia-vida dos neutrófilosécurta e nao se pode esperar a normalizaçao de sua funçao. Um dos grandes problemas é a doença do enxerto versus hospedeiro nas transfusoes de granulócitos, atribuída à contaminaçao do concentrado com linfócitos imunocompetentes. Reaçoes adversas agudas sao frequentes e potencialmente graves, com elevaçao da temperatura do receptor durante ou logo após as transfusoes, principalmente quando a velocidade de transfusao é superior a 2,0 x1010/hora. Outra complicaçao possível é a hemólise das hemáceas do doador, ou mesmo do receptor, se nao for respeitada a compatibilidade ABO; e reaçao adversa aguda causada pela concentraçao dos granulócitos no leito vascular pulmonar, levando à dispneia, infiltrados pulmonares e hipoxemia, principalmente em pacientes aloimunizados.
Os critérios de eficácia do tratamento da neutropenia congênita incluem reduçao das complicaçoes decorrentes da infecçao, número de infecçoes e melhora na qualidade de vida1,6. Casos moderados de neutropenia acompanhados de infecçao leve ou superficial sao tratados com antibióticos orais, ao passo que neutropenia grave e septicemia necessitam de hospitalizaçao. Nesta situaçao sao necessários exames como hemocultura, urinálise, cultura e antibiograma, raios-x de tórax e uso imediato de antibióticos endovenosos, como cefalosporinas de terceira geraçao e aminoglicosídeos. Se a febre persistir por mais de 48 horas, sao indicados antifúngicos6. Em casos graves, é indicado o uso de rHUG-CSF na dose inicial de 5 µg/ kg/dia. O tratamento para correçao da neutropenia consiste no uso de fatores de crescimento hematopoiético (rHUG-CSF, rHUGM-CSF). Na prática, o rHUG-CSF é mais utilizado, pois o rHUGM-CSF é menos eficaz, e menos tolerável (causa sintomas semelhante à gripe e eosinofilia). O rHUG-CSF é encontrado em duas formas: filgrastim (Neupogen 330 µg ou 480 µg) e lenograstin (Granocyte 130 µg ou 340 µg)4,6,8. Estas duas moléculas sao semelhantes, sendo o lenograstin a forma glicosilada do rHUG-CSF, possuindo os mesmos efeitos biológicos, embora alguns estudos apontem maior aumento na produçao de neutrófilos pelo uso de filgrastim108. O esquema de tratamento consiste em duas fases, uma de induçao e outra de manutençao. A fase de induçao é avaliada individualmente, sendo indicado o rHUG-CSF na dose de 5 µg/kg/dia, por via subcutânea, com efeito verificado através do aumento da contagem de neutrófilos (> 1.500 neutrófilos/mm3 de sangue) acompanhado de melhora clínica. Caso a resposta seja rápida ou excessiva (> 5.000 neutrófilos/mm3 de sangue), a dose deve ser reavaliada. Uma vez determinada a dose mímima de induçao, o tratamento de manutençao pode ser estabelecido, sendo monitorado a cada 4 a 6 meses. Dois terços dos pacientes com neutropenia congênita grave respondem à dose diária de 2 a 10 µg/kg; 20% a doses de 10 a 20 µg/kg; e um pequeno número de pacientes a doses superiores a 100 µg/kg4,6,8. Em pacientes com neutropenia cíclica a dose indicada é de 5 µg/kg/dia, e a contagem pode exceder em algumas ocasioes 30.000 neutrófilos/mm3 de sangue6. Quando utilizado em períodos inferiores a 15 dias na dose de 1 a 5 µg/kg/dia em pacientes pediátricos e adultos com câncer que receberam quimioterapia, pode causar sintomas semelhantes à gripe. Dor óssea é descrita em apenas 2 a 5% dos casos, que desaparece rapidamente após interrupçao do tratamento1,6. Em pacientes que necessitam de tratamento prolongado, o rHUG-CSF pode causar monocitose (> 1.500 monócitos/mm3 de sangue), eosinofilia e trombocitopenia, que tende a ser moderada e regride com reduçao da dose4,6,8. A trombocitopenia pode levar à esplenomegalia, principalmente na doença do acúmulo de glicogênio Ib, com ruptura do baço e necessidade de cirurgia emergencial. Pode ocorrer vasculite leucocitoclástica (síndrome de Sweet) em tratamentos com menos de 1 mês de duraçao6. Glomerulonefrite mesangioproliferativa foi descrita, com desaparecimento após reduçao de dose ou interrupçao do tratamento, e osteoporose é vista em um quarto dos pacientes com neutropenia congênita grave109, seguido de fratura patológica.
Existe um aumento do risco de desenvolvimento de leucemiae mielodisplasia em pacientes com neutropenia congênita submetidos ao tratamento com rHUG-CSF com mutaçao em ELANE110, HAX1111, WASP112, SBDS113, G6PC3 ou SLC37A4114, mas ausente em pacientes com neutropenia cíclica. O risco aumenta com uso de altas doses de rHUG-CSF (acima de 15 µg/kg/dia) durante longos períodos115. O monitoramento do risco de leucemia pode ser feito através da contagem rotineira de leucócitos, hemácias, plaquetas, e mielograma. Existe outra forma de rHUG-CSF (PergFilgrastim Neulasta), uma combinaçao de filgrastim com polietilenoglicol, que possui meia-vida de 15 a 80 horas; diminuindo o número de injeçoes, o que por outro lado, possibilita o surgimento de superdosagem, efeitos adversos e perda da eficácia116. Outra possibilidade de tratamento é o transplante de células tronco hematopoiéticas (hematopoietic stem cell transplantation/HSCT), sendo indicado em casos refratários ao tratamento com rHUG-CSF que apresentam infecçoes recorrentes graves, resistência ao tratamento (dose superior a 50 µg/kg/dia), ou pacientes com pancitopenia sem detecçao de mielodisplasia/leucemia1,6,117,118. A terapia gênica é uma estrategia terapêutica promissora para formas variantes de neutropenia congênita, contudo, tais estratégias experimentais ainda necessitam de uma avaliaçao cuidadosa dos potenciais riscos e seus benefícios119-123.
DISCUSSAO
Os neutrófilos sao a populaçao mais abundante de leucócitos no sangue que atuam nas fases iniciais da resposta inflamatória. Neutropenia é definida como reduçao do número absoluto de neutrófilos na circulaçao sanguínea, abaixo de 2.000 neutrófilos/mm3 de sangue em crianças entre 2 a 12 meses de idade, e abaixo de 1.500 neutrófilos/mm3 de sangue em crianças com mais de um ano de idade, sendo considerada grave quando a contagem for inferior a 500 neutrófilos/mm3 de sangue, e crônica quando permanece baixa durante os últimos 3 meses.
Pacientes com neutropenia congênita grave apresentam contagem abaixo de 500 neutrófilos/mm3 de sangue, febre, infecçoes recorrentes e graves da pele, trato respiratório e digestório no início da vida. A suspeita de neutropenia congenita deve ser avaliada através de exames clínicos e laboratoriais seguindo de diagnóstico molecular. A identificaçao dos defeitos moleculares permite o diagnóstico molecular de muitos pacientes, servindo como pré-requisito para uma melhor avaliaçao da evoluçao da doença e riscos associados às terapias. A neutropenia congênita grave corresponde a um grupo genético heterogêneo que afeta a diferenciaçao mieloide para a produçao de neutrófilos. A forma autossômica dominante pode ser causada por mutaçao do gene ELANE e mutaçao do gene CSF3R, ambas consideradas neutropenia congênita sem manifestaçao extra-hematopoiética. Outras formas de neutropenia congênita incluem a mutaçao dos genes GFI1, HAX1, G6PC3 e WAS.
O diagnóstico da neutropenia congênita é suspeito em crianças com infecçao bacteriana ou fúngica persistente associado com neutropenia. A profilaxia consiste no uso de antibióticos de amplo espectro, que aumentam significativamente o tempo de sobrevida dos pacientes. As formas de tratamento para correçao da neutropenia incluem o uso de fatores de crescimento hematopoiético recombinante humano (rHUG-CSF e rHUGM-CSF) e o transplante de células-tronco hematopoiéticas. Existe um aumento do risco de desenvolvimento de leucemia e mielodisplasia em pacientes com neutropenia congênita submetidos ao tratamento com rHUG-CSF. Além disso, formas de terapias gênicas podem fornecer uma fonte curativa poderosa como estratégia terapêutica para formas variantes de neutropenia congênita no futuro próximo. Apesar disso, tais estratégias, experimentais ainda, necessitam de uma avaliaçao cuidadosa dos potenciais riscos e seus benefícios. As neutropenias congênitas representam um modelo natural de estudo da granulopoiese, e inúmeras alteraçoes moleculares responsáveis pela neutropenia nao envolvem somente genes com papel transcricional na granulopoiese, mas também em funçoes do retículo endoplasmático, garantindo a estabilidade dos grânulos, seu tráfico intracelular ou empacotamento de proteínas. Dessa forma, os conhecimentos das bases moleculares das neutropenias congênitas providenciam importantes informaçoes sobre a diferenciaçao mieloide, granulopoiese, assim como com relaçao à busca de novas abordagens terapêuticas.
REFERENCIAS
1. Van den Berg JM, Kuijpers TW. Defects in number and function of neutrophilic granulocytes causing primary imunodeficiency. Eur J Pediatr. 2011;170:1369-76.
2. Smaaland R, Sothem RB, Laerum OD, Abrahamsen JF. Rhythms in human bone marrow and blood cells. Chronobiol Int. 2002;19:101-27.
3. Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825-52.
4. Del Vecchio A,Christensen RD. Neonatal neutropenia: what diagnostic evaluation is needed and when is treatment recommended? Early Huma Dev. 2012;88 Suppl 2:S19-24.
5. Peng H-W, Chou C-F, Liang D-C. Hereditary cyclic neutropenia in the male members of a Chinese family with inverted Y chromosome. Brit J Haemat. 2000;110:438-40.
6. Donadieu J, Fenneteau O, Beaupain B, Mahlaoui N, Chantelot CB. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis. 2011;6:1-28.
7. Skokowa J, Germeschausen M, Zeidler C, Welte K. Severe congenital neutropenia: inheritance and pathophysiology. Curr Opin Hemat 2007;14:22-8.
8. Freedman MH, Bonilla MA, Fier C, Bolyard AA, Scarlata D, Boxer LA, et al. Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000;96:429-36.
9. Ishikawa N, Okada S, Miki M, Shirao K, Kihara H, Tsumura M, et al. Neurodevelopmental abnormalities associated with severe congenital neutropenia due to the R86X mutation in the HAX1 gene. J Med Genet. 2008;45:802-7.
10. Smith BN, Ancliff PJ, Pizzey A, Khwaja A, Linch DC, Gale RE. Homozygous HAX1 mutations in severe congenital neutropenia patients with sporadic disease: a novel mutation in two unrelated British kindreds. Brit J Haemat. 2008;144:762-70.
11. Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, et al. LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced incongenital neutropenia. Nature Med 2006; 12: 1191-7. Note: Erratum: Nature Med. 2006;12:1329.
12. Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. New Eng J Med. 2000;343:1703-14.
13. Dong F, Brynes RK, Tidow N, Welte K,Lowenberg B, Touw IP. Mutations in the gene for the granulocyte colony-stimulating-factor receptor in patients with acute myeloid leukemia preceded by severe congenital neutropenia. New Eng J Med. 1995;333:487-93.
14. Rosenberg PS, Alter BP, Link DC, Stein S, Rodger E, Bolyard AA, et al. Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Brit J Haemat. 2007;140:210-13.
15. Tidow N, Pilz C, Teichmann B, Muller-Brechlin A, Germeshausen M, Kasper B, et al. Clinical relevance of point mutations in the cytoplasmic domain of the granulocyte colony-stimulating factor receptor gene in patients with severe congenital neutropenia. Blood. 1997;89:2369-75.
16. McLemore ML, Poursine-Laurent J, Link DC. Increased granulocyte colony-stimulating factor responsiveness but normal resting granulopoiesis in mice carrying a targeted granulocyte colony-stimulating factor receptor mutation derived from a patient with severe congenital neutropenia. J Clin Invest. 1998;102:483-92.
17. AytekinC, GermeshausenM, Tuygun N, Tanir G, Dogu F, Ikinciogullari A. Eponym. Kostmann disease. Eur J Pediatr. 2010;169:657-60.
18. Aytekin C, Germeshausen M, Tuygun N, Tanir G, Dogu F, Ikinciogullari A. Kostmann disease with developmental delay in three patients. Eur J Pediatr. 2010;169:759-62.
19. Boztug K, Appaswamy G, Ashikov A, Schäffer AA, Salzer U, Diestelhorst J, et al. A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009;360:32-43.
20. Xia J, Bolyard AA, Rodger E, Stein S, Aprinkian AA, Dale DC, et al. Prevalence of mutations in ELANE,GFI1 , HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol. 2009;147:535-42.
21. Devriend K, Kim AS, Mathijs G, Frints SGM, Schwartz M, Vanden Oord JJ, et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nature Genet. 2001;27:313-17.
22. Ancliff PJ, Blundell MP, Cory GO, Calle Y, Worth A, Kempski H, et al. Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006;108:2182-9.
23. Beel K, Cotter MM, Blatny J, Bond J, Lucus G, Green F, Vanduppen V, et al. A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene. Brit J Haemat. 2008;144:120-6.
24. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nature Genet. 2003;34:70-4.
25. Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JRJr. WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000;91:368-76.
26. Liu Q, Chen H, Ojode T, Gao X, Anaya-O'Brien S, Turner NA, et al. WHIM syndrome caused by a single amino acid substitution in the carboxy-tail of chemokine receptor CXCR4. Blood. 2012[Epubahead of print].
27. Palmer SE, Stephens K, Dale DC. Genetics, phenotype, and natural history of autosomal dominant cyclic hematopoiesis. Am J Med Genet. 1996;66:413-22.
28. Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nature Genet. 1999;23:433-6.
29. Morley AA, Carew JP, Baikie AG. Familial cyclical neutropenia. Brit J Haemat. 1967;13:719-38.
30. Krance RA, Spruce WE, Forman SJ, Rosen RB, Hecht T, Hammond WP, et al. Human cyclic neutropenia transferred by allogeneic bone marrow grafting. Blood. 1982;60:1263-6.
31. Dale DC, Ward SB, Kimball HR, Wolff SM. Studies of neutrophil production and turnover in grey collie dogs with cyclic neutropenia. J Clin Invest. 1972;51:2190-6.
32. Lothrop CDJr, Coulson PAJr, Nolan HL, Cole B, Jones JB, Sanders WL. Cyclic hormonogenesis in gray collie dogs: interactions of hematopoietic and endocrine systems. Endocrinology. 1987;120:1027-32.
33. Kuijpers TW, Alders M, Tool ATJ, Mellink C, Roos D, Hennekam RCM. Hematologic abnormalitiesin Shwachman Diamond syndrome: lack of genotype-phenotype relationship. Blood. 2005;106:356-61.
34. Popovic M, Goobie S, Morrison J, Ellis L, Ehtesham N, Richards N, et al. Fine mapping of the locus for Shwachman-Diamond syndrome at 7q11, identification of shared disease haplotypes, and exclusion of TPST1 as a candidate gene. Europ J Hum Genet. 2002;10:250-8.
35. Dror Y, Ginzberg H, Dalal I, Cherepanov V, Downey G, Durie P, et al. Immune function in patients with Shwachman-Diamond syndrome. Brit J Haemat. 2001;114:712-7.
36. Toiviainen-Salo S, Makitie O, Mannerkoski M, Hamalainen J, Valanne L, Autti T. Shwachman-Diamond syndrome is associated with structural brain alterations on MRI. Am J Med Genet. 2008;146A:1558-64.
37. Barth PG, Van't Veer-Korthof ET, Van Delden L, Van Dam K, Van der Harten JJ, Kuipers JRG. An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leukocytes. In: Busch HFM, Jennekens FGI, Schotte HR, eds. Mitochondria and Muscular Diseases. Beetsterzwaag, The Netherlands: Mefar (pub) 1981. p. 161-4.
38. Hodgson S, Child A, Dyson M. Endocardial fibroelastosis: possible X linked inheritance. J Med Genet. 1987;24:210-14.
39. Kelley RI, Clark BJ, Morton DH, Sherwood WG. X-linked cardiomyopathy, neutropenia, and increased urinary levels of 3-methylglutaconic and 2-ethylhydracrylic acids. (Abstract) Am J Hum Genet. 1989;45 (suppl):A7.
40. Orstavik KH, Skjorten F, Hellebostad M, Haga P, Langslet A. Possible X linked congenital mitochondrial cardiomyopathy in three families. J Med Genet. 1993;30:269-72.
41. Barth PG, Valianpour F, Bowen VM, Lam J, Duran M, Vaz FM, et al. X-linked cardioskeletal myopathy and neutropenia(Barthsyndrome): an update. Am J Med Genet. 2004;126A:349-54.
42. Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, et al. A Drosophila model of Barth syndrome. Proc Nat Acad Sci. 2006;103:11584-8.
43. Spritz RA. Multi-organellar disorders of pigmentation: tied up in traffic. Clin Genet. 1999;55:309-17.
44. Huizing M, Scher CD, Strovel E, Fitzpatrick DL, Hartnell LM, Anikster Y, et al. Nonsense mutations in ADTB3A cause complete deficiency of the beta-3A subunit of adaptor complex-3andsevere Hermansky-Pudlak syndrome type 2. Pediat Res. 2002;51:150-8.
45. Jung J, Bohn G, Allroth A, Boztug K, Brandes G, Sandrock I, et al. Identification of a homozygous deletion in the AP3B1 gene causing Hermansky-Pudlak syndrome, type 2. Blood. 2006;108:362-9.
46. Fontana S, Parolini S, Vermi W, Booth S, Gallo F, Donini M, et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood 2006;107:4857-6.
47. Dell'Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky-Pudlak syndrome due to mutations in the beta-3A subunit of the AP-3 adaptor. Molec Cell. 1999;3:11-21.
48. Enders A, Zieger B, Schwarz K, Yoshimi A, Speckmann C, Knoepfle E-M, et al. Lethal hemophagocytic lymphohistiocytosis in Hermansky-Pudlak syndrome type II. Blood. 2006;108:81-7.
49. Sugita M, Cao X, Watts GFM, Rogers RA, Bonifacino JS, Brenner MB. Failure of trafficking and antigen presentation by CD1 in AP-3-deficient cells. Immunity. 2002;16:697-706.
50. Clark RH, Stinchcombe JC, Day A, Blott E, Booth S, Bossi G, et al. Adaptor protein 3-dependent microtubule-mediated movement of lytic granules to the immunological synapse. Nature Immun. 2003;4:1111-20.
51. Fontana S, Parolini S, Vermi W, Booth S, Gallo F, Donini M, et al. Innate immunity defects in Hermansky-Pudlak type 2 syndrome. Blood. 2006;107:4857-64.
52. Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schäffer AA, Ret al. A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007;13:38-45.
53. Mostefai R, Morice-Picard F, Boralevi F, Sautarel M, Lacombe D, Stasia MJ, et al. Poikiloderma with neutropenia, Clericuzio type, in a family from Morocco. Am J Med Genet. 2008;146A:2762-9.
54. Tanaka A, Morice-Picard F, Lacombe D, Nagy N, Hide M, Taieb A, et al. Identification of a homozygous deletion mutation in C16orf57 in a family with Clericuzio-type poikiloderma with neutropenia. Am J Med Genet. 2010;152A:1347-8.
55. Wang LL, Levy ML, Lewis RA, Chintagumpala MM, Lev D, Rogers M, et al. Clinical manifestations in a cohort of 41 Rothmund-Thomson syndrome patients. Am J Med Genet. 2001;102:11-7.
56. Wang LL, Gannavarapu A, Clericuzio CL, Erickson RP, Irvine AD, Plon SE. Absence of RECQL4 mutations in poikiloderma with neutropenia in Navajo and non-Navajo patients. (Letter) Am J Med Genet. 2003;118A:299-301.
57. Concolino D, Roversi G, Muzzi GL, Sestito S, Colombo EA, Volpi L, et al. Clericuzio-type poikiloderma with neutropenia syndrome in three sibs with mutations in the C16orf57 gene: delineation of the phenotype. Am J Med Genet. 2010;152A:2588-94.
58. Senior B, Loridan L. Functional differentiation of glycogenoses of the liver with respect to the use of glycerol. New Eng J Med. 1968;279:965-70.
59. Kure S, Suzuki Y, Matsubara Y, Sakamoto O, Shintaku H, Isshiki G, et al. Molecular analysis of glycogen storage disease type Ib: identification of a prevalent mutation among Japanese patients and assignment of a putative glucose-6-phosphate translocase gene to chromosome 11. Biochem Biophys Res Commun. 1998;248:426-31.
60. Chou JY, Mansfield BC. Molecular genetics of type 1 glycogen storage diseases. Trends Endocr Metab. 1999;10:104-13.
61. Ambruso DR, McCabe ERB, Anderson D, Beaudet A, Ballas LM, Brandt IK, et al. Infectious and bleeding complications in patients with glycogenosis Ib. Am J Dis Child. 1985;139:691-7.
62. Ueno N, Tomita M, Ariga T, Ohkawa M, Nagano S, Takahashi Y, et al. Impaired monocyte function in glycogen storage disease type Ib. Europ J Pediat. 1986;145:312-14.
63. Kuijpers TW, Maianski NA, Tool ATJ, Smit PA, Rake JP, Roos D, et al. Apoptotic neutrophils in the circulation of patients with glycogen storage disease type 1b (GSD1b). Blood. 2003;101:5021-4.
64. Talente GM, Coleman RA, Alter C, Baker L, Brown BI, Cannon RA, et al. Glycogen storage disease in adults. Ann Intern Med. 1994;120:218-26.
65. Roe TF, Coates TD, Thomas DW, Miller JH, Gilsanz V. Treatment of chronic inflammatory bowel disease in glycogen storage disease type Ib with colony-stimulating factors. New Eng J Med. 1992;326:1666-9.
66. Parri V, Katzaki E, Uliana V, Scionti F, Tita R, Artuso R, et al. High frequency of COH1 intragenic deletions and duplications detected by MLPA in patients with Cohen syndrome. Eur J Hum Genet. 2010;18:1133-40.
67. Kolehmainen J, Black GCM, Saarinen A, Chandler K, Clayton-Smith J, Traskelin A-L, et al. Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003;72:1359-69.
68. Kivitie-Kallio S, Summanen P, Raitta C, Norio R. Ophthalmologic findings in Cohen syndrome: along-term follow-up. Ophthalmology. 2000;107:1737-45.
69. Rivera-Brugues N, Albrecht B, Wieczorek D, Schmidt H, Keller T, Gohring I, et al. Cohen syndrome diagnosis using whole genome arrays. J Med Genet. 2011;48:136-40.
70. De Ravel TJL, Dillen K, Fryns JP. A new association of mental retardation, short stature, unusual face, radio-ulnar synostosis and retinal pigment abnormalities: Cohen syndrome with thrombocytopenia. (Letter) Genet Counsel. 2002;13:475-6.
71. Kivitie-Kallio S, Norio R. Cohen syndrome: essential features, natural history, and heterogeneity. Am J Med Genet. 2001;102:125-35.
72. Ku C-L, von Bernuth H, Picard C, Zhang S-Y, Chang H-H, Yang K, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407-22.
73. Picard C, Puel A, Bonnet M, Ku C-L, Bustamante J, Yang K, et al. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003;299:2076-9.
74. Haraguchi S, Day NK, Nelson RPJr, Emmanuel P, Duplantier JE, Christodoulou CS, Good RA. Interleukin 12 deficiency associated with recurrent infections. Proc Nat Acad Sci. 1998;95:13125-9.
75. Hoarau C, Gerard B, Lescanne E, Henry D, Francois S, Lacapere J-J, et al. TLR9 activation induces normal neutrophil responses in a child with IRAK-4 deficiency: involvement of the direct PI3K pathway. J Immun. 2007;179:4754-65.
76. Singh A, Zarember KA, Kuhns DB, Gallin JI. Impaired priming and activation of the neutrophil NADPH oxidase in patients with IRAK4 or NEMO deficiency. J Immun. 2009;182:6410-17.
77. Gallardo E, Claeys KG, Nelis E, Garcia A, Canga A, Combarros O, et al. Magnetic resonance imaging findings of leg musculature in Charcot-Marie-Tooth disease type 2 due to dynamin 2 mutation. J Neurol. 2008;255:986-92.
78. Zuchner S, Noureddine M, Kennerson M, Verhoeven K, Claeys K, De Jonghe P, et al. M. Mutations in the pleckstrin homology domain of dynamin 2 cause dominant intermediate Charcot-Marie-Tooth disease. Nature Genet. 2005;37:289-94.
79. Claeys KG, Zuchner S, Kennerson M, Berciano J, Garcia A, Verhoeven K, et al. Phenotypic spectrum of dynamin 2 mutations in Charcot-Marie-Tooth neuropathy. Brain. 2009;132:1741-52.
80. McKusick VA, Eldridge R, Hostetler JA, Egeland JA, Ruangwit U. Dwarfism in the Amish. II. Cartilage-hair hypoplasia. Bull Johns Hopkins Hosp. 1965;116:285-326.
81. Hirose Y, Nakashima E, Ohashi H, Mochizuki H, Bando Y, Ogata T, et al. Identification of novel RMRP mutations and specific founder haplotypes in Japanese patients with cartilage-hair hypoplasia. J Hum Genet. 2006;51:706-10.
82. Ridanpaa M, Sulisalo T, de la Chapelle A, Kaitila I. Genetic and physical mapping of the cartilage-hair hypoplasia locus on 9p13. (Abstract) Am J Hum Genet. 1995;57:A201.
83. Bonafe L, Schmitt K, Eich G, Giedion A, Superti-Furga A. RMRP gene sequence analysis confirms a cartilage-hair hypoplasia variant with only skeletal manifestations and reveals a high density of single-nucleotide polymorphisms. Clin Genet. 2002;61:146-51.
84. Toiviainen-Salo S, Kajosaari M, Piilonen A, Makitie O. Patients with cartilage-hair hypoplasia have an increased risk for bronchiectasis. J Pediat. 2008;152:422-8.
85. Makitie O, Kaitila I, Savilahti E. Susceptibility to infections and in vitro immune functions in cartilage-hair hypoplasia. Europ J Pediat. 1998;157:816-20.
86. Williams MS, Ettinger RS, Hermanns P, Lee B, Carlsson G, Taskinen M, et al. The natural history of severe anemia in cartilage-hair hypoplasia. Am J Med Genet. 2005;138A:35-40.
87. Fryns JP. Hypersplenism and portal hypertension with vena porta thrombosis in cartilage-hair hypoplasia (metaphyseal chondrodysplasia, McKusick type, MIM *250250). (Letter) Genet Counsel. 2000;11:277-8.
88. MakitieO,KaitilaI.Cartilage-hair-hypoplasia clinical manifestations in 108 Finnish patients. Europ J Pediat. 1993;152:211-7.
89. Bailly-Botuha C, Jaubert F, Taam RA, Galmiche L, Picard C, Bellon G, deBlicJ. Diffuse lymphoplasmacytic brochiolitis in cartilage-hair hypoplasia. J Pediat. 2008;152:429-33.
90. Taskinen M, Ranki A, Pukkala E, Jeskanen L, Kaitila I, Makitie O. Extended follow-upofthe Finnishcartilage-hair hypoplasia cohort confirms high incidence of non-Hodgkin lymphoma and basal cell carcinoma. Am J Med Genet. 2008;146A:2370-5.
91. Arvin AM, Kushner JH, Feldman S, Baehner RL, Hammond D, Merigan TC. Human leukocyte interferon from the treatment of varicella in children with cancer. New Eng J Med. 1982;306:761-5.
92. Wood MJ. Current experience with antiviral therapy for acute herpes zoster. Ann Neurol. 1994;35:S65-S68.
93. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: report ona United States registry of 201 patients. Medicine. 2006;85:193-202.
94. Wan C, Yu HH, Lu MY, Lee JH, Wang LC, Lin YT, et al. Clinical manifestations and outcomes of pediatric chronic neutropenia. J Forms Med Assoc. 2012;111:220-7.
95. Will N, Seger RA, Betzler C, Dockter G, Graf N, Büttner M, et al. Bare lymphocyte syndrome-combined immunodeficiency and neutrophil dysfunction. Eur J Pediatr. 1990;149:700-4.
96. Perreault S, Bernard G, Lortie A, Le Deist F, Decaluwe H. Ataxiatelangiectasia presenting with a novel immunodeficiency. Pediatr Neurol. 2012;46:322-4.
97. Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich syndrome: diagnosis, clinical and laboratory manifestations, and treatment. Biol Blood Marrow Transplant. 2009;15(1 Suppl):84-90.
98. Cosar H, Kahramaner Z, Erdemir A, Kanik A, Turkoglu E, Sutcuoglu S, et al. Reticular dysgenesis in a preterm infant: a case report. Pediatr Hematol Oncol. 2010;27:646-9.
99. Akar NA, Adekile AD. Chromosome 22q11.2 deletion presenting with immune-mediated cytopenias, macrothrombocytopenia and platelet dysfunction. Med Pric Pract. 2007;16:318-20.
100. Bohn G, Welte K, Klein C. Severe congenital neutropenia: new genes explain an old disease. Cuur Opin Rheumatol. 2007;19:644-50.
101. Yamamoto K, Watanabe A, Kakihara T, Tanaka A, Uchiyama M. Hemophagocytic lymphohistiocytosis with preceding neurologic signs and neutrophilia. Pediatr Int. 2000;42:167-9.
102. Da Costa L, Moniz H, Simansour M, Tchernia G, Mohandas N, Leblanc T. Diamond-Blackfan anemia, ribosome and erythropoiesis. Transfus Clin Biol. 2010;17:112-9.
103. Leguit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology. 2010;57:655-70.
104. Watkins D, Rosenblatt DS. Inborn errors of cobalamin absorption and metabolism. Am J Med Genet C Semin Med Genet.2011;157:33-44.
105. Finsterer J. Hematological manifestations of primary mitochondrial disorders. Acta Haematol. 2007;118:88-98.
106. Centola M, Wood G, Frucht DM, Galon J, Aringer M, Farrell C, et al. The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood. 2000;95:3223-31.
107. Ulinski T, Aoun B, Toubiana J, Vitkevic R, Bensman A, Donadieu J. Neutropenia in congenital nephrotic syndrome of the Finnish type: role of urinary ceruloplasmin loss. Blood. 2009;113:4820-1.
108. Carlson G, Ahlin A, Dahilof G, Elinder G, Henter J, Paimblad J. Efficacy and safety of two different rG-CSF preparation in the treatment of patients with severe congenital neutropenia. Br J Haematol. 2004;126:127-32.
109. Yakisan E, Sching E, Zeidler C, Bishop NJ, Reiter A, Hirt A, et al. High incidence of significant bone loss inpatients with severe congenital neutroipenia (Kostmann's syndrome). J Pediatrics. 1997;131:592-7.
110. Rosemberg PS, Alter BP, Link DC, Stein S, Rodger E, Bolyand AA, et al. Neutropil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol. 2008;140:210-3.
111. Yetgin S, Olcay L, Koc A, Gemmeshausen M. Transformation of severe congenital neutropenia to early acute lymphoblastic leukemia in a patient with HAX1 mutation and without G-CSF administration or receptor mutation. Leukemia. 2008;22:1797.
112. Beel K, Vandenberghe P. G-CSF receptor (CSF3R) mutations in X-linked neutropenia evolving to acute myeloid leukemia or myelodysplasia. Haematologia. 2009;94:1449-52.
113. Dror Y. Schwachman-Diamond syndrome. Pediatr Blood Cancer. 2005;45:892-901.
114. Pinsk M, Burzynski J, Yhap M, Fraser RB, Cummings B, Ste-Marie M. Acute myelogenous leukemia and glycogen storage disease 1b. J Pediatr Hematol Oncol. 2002;24:756-58.
115. Donadieu J, Leblanc T, Bader MB, Barkaoui M, Fenneteau O, Bertrand Y, et al. Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience on the French Severe Chronic Neutropenia Study Group. Haematologica. 2005;90:45-53.
116. Fioredda F, Calvillo M, Lanciotti M, Lanza T, Giunti L, Castagnola E, et al. Pegfilgrastim in children with severe congenital neutropenia. Pediatr Blood Cancer. 2010;54:465-7.
117. Leguit RJ, van den Tweel JG. The pathology of bone marrow failure. Histopathology. 2010;57:655-70.
118. Connelly JA, Choi SW, Levine JE. Hematopoietic stem cell transplantation for severe congenital neutropenia. Curr Opin Hematol. 2012;19:44-51.
119. Fischer A, Hacein-Bey-Abina S, Cavazzana-Calvo M. Gene therapy for primary immunodeficiencies. Hematol Oncol Clin North Am. 2011;25:89-100.
120. Booth C, Gaspar HB, Thrasher AJ. Gene therapy for primary immunodeficiency. Curr Opin Pediatr. 2011;23:659-66.
121. Porteus M. Homologous recombination-based gene therapy for the primary immunodeficiencies. Ann N Y Acad Sci. 2011;1246:131-40.
122. Rivat C, Santilli G, Gaspar HB, Thrasher AJ. Gene therapy for primary immuno deficiencies. Hum Gene Ther. 2012;23:668-75.
123. Ginn SL, Alexander IE. Genetherapy: Progress in childhood disease. J Paediatr Child Health. 2012;48:466-71.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888