Número Atual: Janeiro-Março 2019 - Volume 3 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Síndromes MonoMAC e Emberger em paciente com mutação no gene GATA2
MonoMAC and Emberger syndromes in a patient with GATA2 mutation
Mariana Jobim1; Enilis Lima Abreu1; Jacinta Bustamante2; Carmen Oleaga2; Tiago Severo Garcia3; Luiz Jobim1,4
DOI: 10.5935/2526-5393.20190016
1. Hospital de Clínicas de Porto Alegre, Serviço de Alergia e Imunologia - Porto Alegre, RS, Brasil
2. Instituto Nacional de Saúde (Inserm), Laboratório de Genética das Doenças Infecciosas - Paris, França
3. Universidade Federal do Rio Grande do Sul, Professor Adjunto de Radiologia - Porto Alegre, RS, Brasil
4. Universidade Federal do Rio Grande do Sul, Professor Titular de Medicina - Porto Alegre, RS, Brasil
Endereço para correspondência:
Luiz Jobim
E-mail: ljobim@hcpa.edu.br
Submetido em: 25/01/2019
Aceito em: 28/02/2019
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
As mutações que ocorrem no gene GATA2 podem ocasionar um amplo espectro de doenças genéticas. Os pacientes podem ter anormalidades na hematopoiese, na linfangiogenesis e na resposta imunológica. Os fenótipos incluem algumas síndromes caracterizadas por monocitopenia e infecção por micobactéria (síndrome MonoMac), síndrome mielodisplásica familiar, leucemia mieloide crônica ou aguda, síndrome de Emberger (linfedema primário), e mais raramente neutropenia, anemia aplástica e deficiência isolada de células NK. A idade da apresentação clínica pode variar desde a infância até a idade adulta. A deficiência autossômica dominante de GATA2 pode permanecer clinicamente silenciosa por décadas, ou mesmo durante toda a vida. Descrevemos o caso de uma jovem brasileira que apresentou a maioria dos problemas ligados à mutação no gene GATA2, observando-se as duas síndromes: MonoMAC e Emberger.
Descritores: Fator de transcrição GATA2, monócitos, infecções por Mycobacterium.
O gene GATA2 (guanina-adenina-timina-adenina 2) é importante membro da família de fatores de transcrição com papel de destaque no desenvolvimento vascular e na hematopoiese. Desde 2011, conhecemos que mutações nesse gene, especialmente nos seus dois domínios com “zink fingers”, possibilitam síndromes como Monocytopenia and Mycbaterium avium Complex (MAC) Infection Syndrome, abreviada como MonoMAC1, síndrome de Emberger2,3, síndrome Mielodisplásica familiar e leucemia mieloide aguda4. A mutação pode acarretar alteração imunológica como citopenia de linfócitos B, de células NK e de monócitos, assim como quadro clínico associado de anemia aplástica, proteinose pulmonar, alterações dermatológicas e autoimunes. A proteinose alveolar acontece devido à redução da capacidade fagocitária dos macrófagos alveolares em eliminar o surfactante. Complementando, um zink finger é uma parte estrutural de uma proteína com dois ou mais íons zinco (Zn2+). Eles estabilizam a molécula proteica e são importantes na regulação de genes.
A primeira publicação sobre as síndromes foi de autoria de Steven Holland do NIH-USA, anteriormente ao conhecimento do gene mutante GATA25. Ele descreveu 18 paciente com susceptibilidade autossômica dominante a micobactérias, fungos e papiloma vírus, associada à monocitopenia. A doença não estava relacionada ao HIV e iniciava, na maioria dos casos, na adolescência com a característica de monocitopenia acentuada, assim como linfopenia B e NK. Dez pacientes apresentaram malignidades como leucemia mieloide aguda/mielodisplasia, carcinoma vulvar e melanoma metastático, leiomiosarcoma com presença do vírus Epstein Barr. Cinco pacientes desenvolveram proteinose alveolar. Embora a maioria dos pacientes apresentasse linfopenia B, a concentração de imunoglobulinas era normal. Neutropenia foi observada em cinco pacientes, sendo que os linfócitos T estavam diminuídos em dez. Em nove deles, o CD4 foi menor do que 300 células/µL. Paniculite com múltiplos nódulos inflamatórios e sensíveis nas extremidades distais, que lembravam eritema nodoso aconteceram em seis pacientes. Outras manifestações cutâneas foram pápulas e placas eritematosas, dolorosas e que ocasionalmente eram acompanhadas de febre e artralgias. Histologicamente, a pele apresentava-se com infiltrado inflamatório.
A seguir, em 2011, o gene GATA2 foi reportado como causa do MonoMAC1, assim como a citopenia de linfócitos B, de células NK e de monócitos, enquanto a susceptibilidade à síndrome de Emberger (linfedema primário) e a leucemia mieloide aguda/mielodisplasia foram também descritas nessa época.
Em 2014, Steven Holland e colaboradores descreveram 57 pacientes do NIH com a deficiência do gene GATA2, ocasionando uma desordem da hematopoiese, dos linfáticos e da imunidade6. Nessa casuística observaram no lado hematológico que a mielodisplasia acontece em 84% dos pacientes e a leucemia mieloide aguda em 14%. As infecções virais severas participaram de 70% dos casos, enquanto a micobacteriose disseminada de 53% e as infecções fúngicas invasivas de 16%. A proteinose alveolar foi identificada em 18% dos pacientes, verrugas em 53%, paniculite em 30%, linfedema em 11%, surdez em 76% e abortos em 33%.
A variabilidade da apresentação clínica foi demonstrada pela análise de uma família com três gerações de indivíduos afetados7. Os membros dessa família apresentaram pancitopenia discreta, mielodisplasia, linfoma não Hodgkin, candidíase muco-cutânea, síndrome de Emberger e leucemia mieloide aguda. A idade de apresentação da doença foi de 14 a 74 anos, ilustrando a necessidade de avaliação genética para a escolha de doadores familiares. A mutação do GATA2 é a causa de imunodeficiência e de neoplasias em adultos. A Síndrome de Emberger é uma doença rara, estimada existir em menos de 1 caso/ milhão de pessoas. Apresenta-se com linfedema primário (com ou sem celulite), principalmente nos membros inferiores (uni ou bilateral); anormalidades linfáticas genitais (linfedema, linfangiectasias, hidrocele); mielodisplasia/leucemia mieloide aguda (CD4/CD8 baixos); surdez. Verrugas múltiplas podem também ocorrer.
Uma recente publicação descreveu 53 famílias com 79 pacientes franceses e belgas8, correspondendo a maior série de indivíduos afetados com essa mutação dentre os aproximadamente 300 pacientes reportados na literatura. A média do início de sintomas foi de 18,6 anos (média de 0-61 anos). As principais manifestações no inicio dos sintomas estavam relacionadas a doenças infecciosas severas (micobactéria, fungos e papiloma vírus) e a malignidades hematológicas. Foram também identificadas novas alterações como leucemia linfoblástica aguda, leucemia mielomonocítica juvenil, leucoencefalopatia multifocal fatal progressiva (JC vírus) e doenças inflamatórias intestinais. Quatorze pacientes já faziam parte do registro de Neutropenia Crônica Severa. A mortalidade foi alta (35% aos 40 anos) e o transplante de medula óssea foi considerada a única terapêutica definitiva e o melhor tratamento. O momento de realizar o transplante é de difícil decisão, mas quanto mais precoce, melhores são os resultados.
RELATO DO CASO
Uma paciente branca de 36 anos foi encaminhada ao nosso Serviço com anemia, febre vespertina, sudorese noturna e emagrecimento desde os 28 anos de idade. Primeira internação em julho de 2012 com diagnóstico de micobacteriose disseminada, não bacilífera e linfopenia. Na hemocultura cresceu Micobacterium kansasii, sendo que um PCR em biópsia do lobo superior direito, guiada por tomografia, identificou Mycobacterium spp.
Paralelamente apresentava linfedema e nódulos inflamatórios nos membros inferiores, sugestivos de eritema nodoso. Importante linfedema inflamatório também ocorreu nas mãos (Figura 1), assim como papilomatose genital. Várias biópsias de pele resultaram em demonstrar inflamação crônica supurativa em derma profundo e tecido adiposo, sem presença de bactérias e fungos.
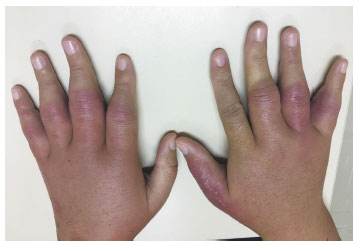
Figura 1 Linfedema das mãos
Recentemente iniciou com febre, dispneia, prostração e intenso angioedema inflamatório nas mãos. Pensamos tratar-se de artrite, mas uma ecografia afastou essa possibilidade, indicando intenso edema. Uma tomografia demonstrou piora do quadro pulmonar, tendo sido internada para avaliação (Figura 2).
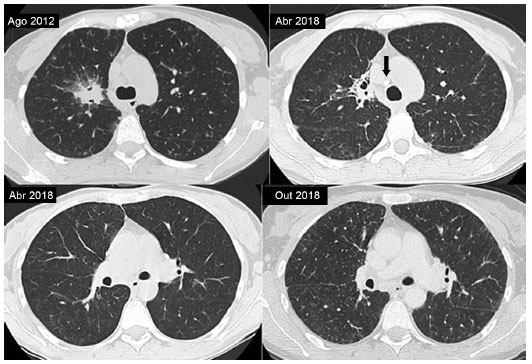
Figura 2 Comparação entre as tomografias computadorizadas de agosto de 2012, abril de 2018 e outubro de 2018. Observa-se em agosto de 2012, consolidação no lobo superior direito e a respectiva regressão em abril de 2018 (retração fibroatelectásica residual) e surgimento de linfonodo calcificado residual no mediastino (seta preta). Em outubro de 2018 aconteceu a progressão dos nódulos centrolobulares no segmento superior dos lobos inferiores
Na história familiar existiram duas irmãs falecidas devido à tuberculose; duas sobrinhas falecidas com mielodisplasia e leucemia mieloide aguda (Figura 3). Resgatamos poucos exames de sangue de uma das irmãs falecidas e constatamos monocitopenia.
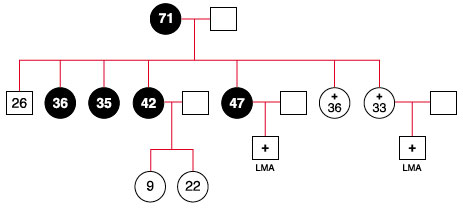
Figura 3 Genealogia da família onde as mulheres com a mutação estão grafadas em preto. A paciente tem 36 anos, sendo que as duas irmãs falecidas foram por tuberculose, e duas sobrinhas por leucemia mieloide aguda (LMA)
Na citometria de fluxo identificamos diminuição do número de linfócitos CD3 circulantes (513/µL – média de 800 a 2.000/µL). Os linfócitos B estavam muito diminuídos (0,1% ou 5/µL – média de 200 a 500/µL), enquanto as células NK, os linfócitos CD4 e CD8 estavam discretamente reduzidos. Foi observada deficiência de IgA circulante (13 mg/dL – média de 150 a 400 mg/dL), assim como a ausência de monócitos.
O sequenciamento dos exons de gene GATA2 foi realizado pelo método de Sanger, identificando os sete exons de codificação e as regiões que flanqueiam os introns. Uma missense mutation foi encontrada c.1192C>T, com alteração ao nível proteico em heterozigose p.Arg398Trp/WT. Além da paciente, sua mãe, duas irmãs e uma sobrinha apresentam a mesma mutação, entretanto não desenvolveram doenças. O único membro da família sem a mutação foi um irmão.
A paciente foi tratada para a micobacteriose por dois anos, passando outros dois anos sem piorar da manifestação pulmonar infecciosa. Devido às queixas de linfedema inflamatório dolorido e a arrastada infecção pulmonar, foi introduzido tratamento com interferon gama-1b (100 µg a cada dois dias durante 60 dias), sem obtermos vantagem clínica ou laboratorial.
A lesão pulmonar aparentemente não foi curada, sendo que deverá ser tratada novamente com tuberculostáticos.
DISCUSSÃO
A mutação germinativa em GATA2 pode ocasionar diversas situações, como a Síndrome MonoMAC, Síndrome de Emberger, imunodeficiência de linfócitos B, células NK, monocitopenia, mielodisplasia e leucemia mieloide aguda e crônica. Outras patologias também já foram descritas nesses pacientes.
A linfopenia de linfócitos B ou células NK ou monocitopenia acontece em 75% dos pacientes6. As infecções virais com papilomatose e micobacterioses não tuberculosa são muito frequentes, assim como o linfedema primário com ou sem reação inflamatória que sugere eritema nodoso. Aliás, na primeira descrição do linfedema de membros inferiores estavam presentes essas lesões assépticas inflamatórias. Nossa paciente também as apresentou nos membros inferiores e nas mãos. Aparentemente o linfedema pode existir com ou sem inflamação, dependendo da existência ou não de micobacteriose, sendo conhecida a alteração ou insuficiência das valvas linfáticas pela mutação no GATA2.
Apresentamos uma paciente adulta com história de infecção crônica com micobacteriose não tuberculosa, assim como linfedema de membros superiores e mãos, deficiência de linfócitos B, deficiência de IgA e ausência de monócitos. Na realidade, ela apresenta uma combinação da síndrome MonoMAC com a síndrome de Emberger, faltando a disfunção hematológica (mielodisplasia ou leucemia mieloide aguda) e surdez para completar o quadro clínico da última síndrome.
Aparentemente, na deficiência de GATA2 observamos sinais e sintomas de mais de uma patologia, dependendo onde a mutação afetar mais o organismo, sendo que provavelmente podemos esperar que outras manifestações clínicas possam ocorrer, dentre as anteriormente expostas, incluindo outras formas de tumores.
As mutações conhecidas são muitas, sendo que a existente na paciente é a mais conhecida até o momento. O sequenciamento realizado foi importante para o diagnóstico definitivo, mas lembramos de que a doença foi inicialmente descrita sem o conhecimento do gene mutante. O quadro de infecção grave por micobacteriose ou papiloma vírus, linfoedema primário e monocitopenia pode ser facilmente identificado clinicamente.
O transplante de células tronco hematopoiéticas é o único tratamento definitivo para os pacientes com deficiência de GATA2. A sobrevida de 54% dos pacientes transplantados faz com que seja difícil sua indicação, mas não fosse isso seria indicada a todos8. O uso de interferon gama-1b não altera o quadro clínico.
Infelizmente a possibilidade de um transplante ficou mais distante pela falta de compatibilidade HLA entre a paciente e seus familiares. Será necessário um transplante com doador não relacionado do REDOME (registro de doadores de medula óssea). A pesquisa nesse banco de dados também não identificou doador compatível adequado, além de existir dificuldade de execução de transplante em paciente cronicamente infectado com micobactérias. O tratamento sintomático está sendo realizado, enquanto não for encontrado um doador de medula óssea ideal, sendo essa a situação dramática de muitos pacientes com doenças genéticas graves.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888