Número Atual: Julho-Setembro 2017 - Volume 1 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Síndrome de desregulação imune, poliendocrinopatia e enteropatia ligada ao X (IPEX): a importância da história familiar para o diagnóstico precoce
Immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome: the importance of family history for early diagnosis
Iramirton Figuerêdo Moreira1; Bruna de Sá Duarte Auto2; Juciene de Matos Braz3; Helena Vieira de Souza Rodrigues4; Thaylla Soares Rodrigues4
DOI: 10.5935/2526-5393.20170044
1. Docente da Faculdade de Medicina da Universidade Federal de Alagoas
2. Médica Residente em Pediatria do Hospital Professor Alberto Antunes - Universidade Federal de Alagoas
3. Acadêmicas de Enfermagem da Faculdade Faculdade Sao Vicente
Endereço para correspondência:
Bruna de Sá Duarte Auto
E-mail: brunaduarte100@gmail.com
Submetido em: 29/06/2017
Aceito em: 25/07/2017.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
A síndrome de desregulaçao imune, poliendocrinopatia e enteropatia ligada ao X (IPEX) é uma síndrome de imunodeficiência primária rara, de herança recessiva, que afeta lactentes do sexo masculino. A doença cursa com enteropatia perdedora de proteínas, dermatite eczematosa e poliendocrinopatias, podendo ser fatal naqueles sem tratamento apropriado. O objetivo deste relato é descrever um caso de IPEX, enfatizando a importância da história familiar para o diagnóstico precoce. O caso descreve um lactente com tipo grave da síndrome, com apresentaçao clínica precoce e história familiar característica, com episódios de morte prematura em doze homens pertencentes à linhagem materna. O diagnóstico por mapeamento genético demostrando mutaçao no gene FOXP3 foi obtido após o óbito do paciente, decorrente de choque séptico. O transplante de células-tronco hematopoiéticas é o melhor tratamento disponível, e na sua ausência, a síndrome IPEX pode ser fatal nos primeiros dois anos de vida. Assim, assegurar um diagnóstico precoce é fundamental.
Descritores: Síndrome de imunodeficiência, doenças genéticas ligadas ao cromossomo X, diagnóstico precoce, dermatite, poliendocrinopatias autoimunes, enteropatias perdedoras de proteínas.
INTRODUÇAO
A síndrome de desregulaçao imune, poliendocrinopatia e enteropatia ligada ao X (IPEX) foi descrita pela primeira vez em 1982 por Powell e cols., em uma grande família com 19 homens afetados em cinco geraçoes, com diarreia letal na maioria dos meninos nos primeiros meses ou anos da vida1. É uma síndrome hereditária rara que afeta lactentes do sexo masculino, decorre de mutaçoes no gene forkhead box protein 3 (FOXP3), localizado no cromossomo Xp11.23 2. Este cromossomo codifica um fator de transcriçao que regula o desenvolvimento das células T CD4+ CD25+, células T reguladoras (Treg) no timo, que estao envolvidas na homeostase imune e na proteçao contra autoimunidade2. A doença cursa com uma enteropatia perdedora de proteínas, diarreia refratária, dermatite eczematosa crônica, poliendocrinopatias, níveis elevados de imunoglobulina E total (IgE total) e, geralmente, com a presença de autoanticorpos3. Pode ainda cursar com citopenia, nefropatias, hepatopatias, níveis elevados de imunoglobulina A (IgA), alergias alimentares e infecçoes graves recorrentes4,5. De forma geral, a apresentaçao clínica e o prognóstico de pacientes acometidos sao variáveis6. Usualmente, ocorre em lactentes jovens e crianças, podendo ser fatal naqueles sem tratamento apropriado5. O objetivo deste relato é descrever um caso de IPEX, enfatizando a importância da história familiar para o diagnóstico precoce.
RELATO DE CASO
AMVR, masculino, 2 meses de idade, natural e procedente de Paulo Afonso, BA, encaminhado ao Serviço de Pediatria do Hospital Universitário Professor Alberto Antunes por apresentar xerose cutânea desde o nascimento, que evoluiu com alopecia, lesoes papulares eritematosas e pruriginosas, resistentes ao tratamento tópico com corticoesteroides e hidratantes, sendo diagnosticado inicialmente como dermatite do tipo ictiosiforme (Figura 1). Antecedentes pessoais: nascido de parto operatório com polidramnia, a termo, com peso de 3.790 gramas e comprimento de 54 centímetros, pré-natal sem intercorrências, calendário vacinal atualizado para idade, sem relato de episódios de reaçao vacinal e teste de triagem neonatal master sem alteraçoes. Antecedentes familiares: pai e mae ambos com 24 anos de idade, nao consanguíneos, com uma filha de 6 anos de idade, todos saudáveis. Bisavós maternos consanguíneos (primos de 1° grau) e história de 12 óbitos em lactentes jovens do sexo masculino com quadro de diarreia e lesoes de pele, conforme heredograma familiar (Figura 2). Exames laboratoriais admissionais mostravam anemia normocrômica e normocítica, plaquetose e contagem de leucócitos dentro da normalidade, com predomínio de linfócitos, hipomagnesemia e hipoalbuminemia. Nos primeiros dias do internamento, evidenciou-se aumento progressivo da contagem de leucócitos (24.100 leucócitos), com diminuiçao dos neutrófilos (1.687 neutrófilos) e eosinofilia importante (8.676 eosinófilos), quando foi levantada a hipótese de imunodeficiência primária e realizados exames para investigaçao com linfócitos T CD3 (3.393/mm3 ; Percentil 50), CD4 (2.782/mm3; Percentil 50) e CD8 (617/mm3; Percentil 10); linfócitos B CD19 (799/mm3; Percentil 10), IgE total (2.926 KU/L;VR:até 8,6 KU/L), IgG (83 mg/dL; Percentil 3), IgA (16 mg/dL; Percentil 50), IgM (28 mg/dL; Percentil 3) e glicemia normal (56 mg/dL). Com a introduçao da fórmula infantil na dieta, o paciente iniciou quadro de diarreia, evoluindo com desidrataçao, piora das lesoes cutâneas e edema das extremidades. Nesse momento, iniciou-se a investigaçao da enteropatia e alergia à proteína do leite de vaca, sendo iniciada dieta com fórmula infantil de aminoácidos livres. Pesquisa de sangue oculta em fezes positiva e a dosagem de calprotectina fecal elevada (943 µg/g; VR: ≤ 200 µg/g). Os demais exames: p-ANCA, c-ASCA, pesquisa de leucócitos fecais, alfa 1-antitripsina fecal, clostridium difficile toxinas A e B, gordura fecal e dosagem de fator anti-nuclear (FAN) dentro da normalidade. O mapeamento genético evidenciou uma variante patogênica no gene FOXP3 (c.1150G>A, p.A384T) definindo o diagnóstico de síndrome IPEX. No entanto, o resultado só foi obtido após o óbito do paciente, decorrente de choque séptico.
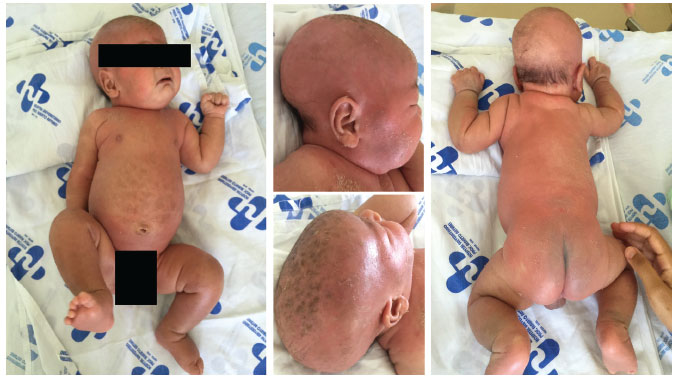
Figura 1 Criança do sexo masculino, com 2 meses de idade, com xerose, alopecia e lesoes papulares eritematosas e pruriginosas
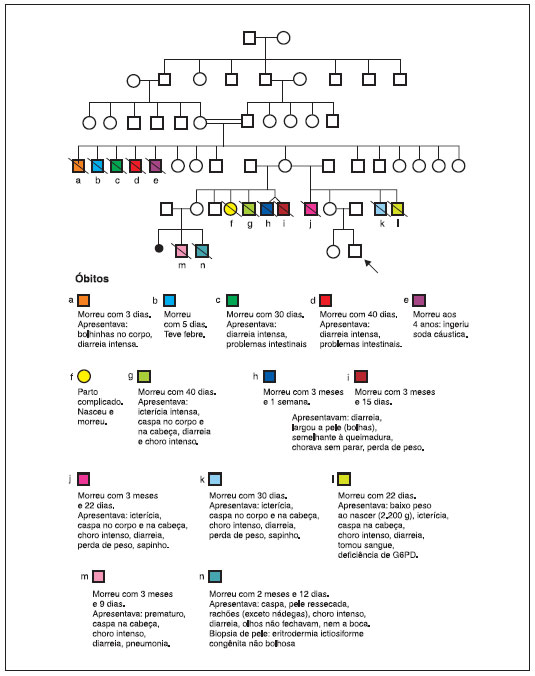
Figura 2 Heredograma do paciente
DISCUSSAO
As imunodeficiências primárias (IDP) ligadas ao X sao doenças que, tipicamente, nao afetam as mulheres ou causam efeitos menos graves que nos homens. Em geral, essas doenças sao diagnosticadas durante a primeira infância, e recém-nascidos do sexo masculino afetados tendem a sofrer de infecçoes bacterianas, fúngicas e virais recorrentes, que podem resultar em septicemia grave e, eventualmente, levar à morte prematura. Isso porque o cromossomo X tem uma grande influência na formaçao da imunidade em seres humanos7. A maioria dessas IDPs ligadas ao X sao caracterizadas por uma ausência ou diminuiçao do número de células imunes, pela presença de células imunes nao funcionais ou mesmo pela diminuiçao da sinalizaçao mediada por citocinas, o que pode agravar o desfecho clínico dos pacientes afetados7.
Este caso descreve um tipo grave da síndrome IPEX, com apresentaçao clínica precoce de dermatite e enteropatia autoimune, além dos quadros de infecçao. A história familiar do caso é característica, com episódios de morte prematura ou achados clínicos similares em homens pertencentes à linhagem materna, enquanto as mulheres sao saudáveis1. A maioria dos pacientes portadores da síndrome nasce após uma gravidez sem intercorrências de pais nao consanguíneos, a termo, com peso e comprimento adequados para idade gestacional1,8. A história familiar cuidadosa pode revelar a presença de indivíduos do sexo masculino na linhagem materna com fenótipo clínico semelhante, morte precoce ou abortos espontâneos múltiplos8. O aparecimento das manifestaçoes clínicas da IPEX geralmente ocorre nos primeiros meses, mas em alguns casos nos primeiros dias ou semanas de vida, e pode ser rapidamente fatal, se nao diagnosticado e tratado adequadamente8. Os casos mais graves sao caracterizados pelo início precoce de uma tríade de manifestaçoes clínicas: diarreia secundária à enteropatia autoimune, Diabetes mellitus tipo 1 (DM1) e eczema8. Podemos observar outras manifestaçoes tais como: citopenias, doenças renais, hepática, alergias alimentares com níveis extremamente elevados de IgE e uma intensa eosinofilia periférica9. A enteropatia cursa com uma diarreia aquosa ou mucoide-sanguinolenta. Histologicamente, é caracterizada por atrofia das vilosidades em duodeno e inflamaçao com destruiçao glandular afetando todas as partes do sistema digestivo, o que pode levar a má absorçao e consequente déficit no crescimento6. A DM 1 ocorre secundariamente à destruiçao das ilhotas pancreáticas por infiltraçao de células T6, podendo manifestar-se antes ou após o aparecimento da enterite10.
No presente caso, os valores glicêmicos apresentaram-se dentro da normalidade, e nao foi diagnosticado DM1. Apesar da DM1 fazer parte da tríade das manifestaçoes clínicas típicas da síndrome IPEX, o estudo realizado por Barzaghi e col.8 mostrou que 35% dos 136 pacientes com a síndrome nao apresentavam DM1. Segundo Barzaghi e col.8, as manifestaçoes cutâneas aparecem nos primeiros meses de vida e podem ser o primeiro sinal da doença. Em lactentes, a dermatite tem sido descrita como eczematosa, psoriasiforme ou ictiosiforme, mas também foram relatados casos de eritroderma esfoliativa. Em crianças mais velhas, outros achados incluem urticária intermitente, alopecia universal, onicodistrofia, penfigoide nodular e penfigoide bolhoso na maioria dos pacientes1 1. O prurido intenso e de difícil controle com anti-histamínicos é uma manifestaçao frequente nestes pacientes. As lesoes cutâneas normalmente sao resistentes aos tratamentos clássicos com corticosteroides tópicos ou com tacrolimus, e sao susceptíveis a infecçoes bacterianas com potencial desenvolvimento de sepse8, uma das principais causas de morte nos pacientes com IPEX, juntamente com complicaçoes da DM1 e diarreia intratável12. Nos pacientes que apresentam sepse, os agentes mais comumente identificados sao os enterococos, estafilococos, cândida e citomegalovírus6. Neste caso em estudo, nao foram identificados agentes patogênicos em culturas de sangue e urina. Porém, em quadros de sepse, apenas cerca de 30% dos casos apresentam hemocultura positiva, e em outros 30% a identificaçao é possível por meio de culturas de outros sítios13. Além disso, a sensibilidade e a especificidade das hemoculturas podem ser afetadas pela técnica de coleta, gerando resultados falsopositivos ou falso-negativos13. Alguns pacientes podem desenvolver outras manifestaçoes, como alopecia, anemia hemolítica, trombocitopenia e hepatite. Em geral, os órgaos afetados apresentam infiltrado linfocitário, com ou sem a presença de autoanticorpos6.
Outros achados laboratoriais típicos da síndrome incluem elevaçao dos níveis de IgE e eosinofilia periférica. Em geral, os níveis séricos de IgA, IgG e IgM sao normais ou baixos pela perda proteica secundária à enteropatia8, como observamos no presente caso. A mutaçao no gene FOXP3 identificada em nosso caso (c.1150G>A) leva a manifestaçoes grave da síndrome IPEX, com morte precoce, porém a mesma mutaçao já foi relatada em pacientes que sobreviveram por mais de 10 anos de idade8. Pelo menos, setenta mutaçoes distintas no gene FOXP3 já foram descritas, e mutaçoes idênticas podem causar fenótipos diferentes1.
A abordagem terapêutica da síndrome IPEX atualmente nao é padronizada, porque o número de casos relatados é limitado e faltam estudos multicêntricos que comparem diferentes estratégias1. Assim, o manejo desses pacientes é baseado em experiências isoladas1.
Os tratamentos atuais disponíveis para pacientes com IPEX incluem terapia de suporte, terapia imunossupressora e transplante de células-tronco hematopoiéticas (TCTH). TCTH alogênico é o melhor tratamento até agora disponível. Para os pacientes que nao se submetem ao transplante, o tratamento é limitado a terapias de suporte, incluindo suporte nutricional e terapia de reposiçao para doenças endócrinas, e a combinaçao de múltiplas drogas imunossupressoras, sem controle permanente de autoimunidade na maioria dos pacientes14. Porém, a única terapia curativa para a síndrome IPEX é o transplante de células tronco hematopoiéticas alogênicas1,14,15. Quanto mais precoce for realizado, ainda no estágio inicial da doença, antes da progressao de lesoes autoimunes, melhor o resultado1,14,15. Na ausência de tratamento curativo com transplante de células-tronco hematopoiéticas, a síndrome IPEX pode ser fatal nos primeiros 2 anos de vida sem tratamento imunossupressor. A disfunçao imunológica profunda que afeta os múltiplos sistemas de órgaos antes do transplante coloca os pacientes em alto risco de complicaçoes relacionadas ao transplante14,15. Apesar disso, os relatos de casos publicados e os resultados de estudos mostraram resultados promissores e melhoria ou resoluçao completa dos sintomas da doença15. Neste sentido, assegurar um diagnóstico precoce é fundamental1.
CONCLUSAO
Este estudo demonstra a importância da história familiar na síndrome de desregulaçao imune, poliendocrinopatia, enteropatia ligada ao X, visto que conhecendo a história poderemos realizar estudos genéticos direcionados para o diagnóstico antes do surgimento das manifestaçoes clínicas. As características clínicas, laboratoriais e o reconhecimento oportuno da doença leva a benefícios terapêuticos significativos. Porém, ainda sao necessários mais estudos, a fim de compreender melhor os fatores que influenciam o desfecho e identificar novas metas terapêuticas.
REFERENCIAS
1. Bacchetta R, Federica B, Roncarolo M. From IPEX syndrome to FOXP3 mutation: a lesson on immune dysregulation. Ann N Y Acad Sci. 2016:1-18.
2. Eujin P, Hye JC, Jae IS, Lim BJ, Jeong HJ, Lee KB, et al. Familial IPEX syndrome: Different glomerulopathy in two siblings. Pediatr Int. 2015;57(2):e59-61.
3. Zennaro D, Scala E, Pomponi D, Caprini E, Arcelli D. Proteomics plus genomics approaches in primary immunodeficiency: the case of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Clin Exp Immunol. 2012;167(1):120-8.
4. Sabina B, Sheilagh MM, Stephen EG, Schneider LC, Lee PY, Notarangelo LD, et al. Immune dysregulation polyendocrinopathy, enteropathy, X-linked syndrome associated with neonatal epidermolysis bullosa acquisita. Pediatr Dermatol. 2015;32(3):e74-7.
5. Chen CA, Chung WC, Chiou YY, Yang YJ, Lin YC, Ochs HD, et al. Quantitative analysis of tissue inflammation and responses to treatment in immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome, and review of literature. J Microbiol Immunol Infect. 2016;49(5):775-82.
6. Bin Dhuban K, Piccirillo CA. The immunological and genetic basis of immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome. Curr Opin Allergy Clin Immunol. 2015;15(6):525-32.
7. Libert C, Dejager L and Pinheiro I. The X chromosome in immune functions: when a chromosome makes the difference. Nat Rev Immunol. 2010;10(8):594-604.
8. Barzaghi F, Passerini L, Bacchetta R. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome: a paradigm of immunodeficiency with autoimmunity. Front Immunol. 2012;3:211.
9. Verbsky JW, Chatila TA. Immune Dysregulation, Polyendocrinopathy, Enteropathy, Xlinked (IPEX) and IPEX-Related Disorders: an Evolving Web of Heritable Autoimmune Diseases. Curr Opin Pediatr. 2013;25(6):708-14.
10. Zama D, Cocchi I, Masetti R, Specchia F, Alvisi P, Gambineri E, et al. Late onset of immunodysregulation, polyendocrinopathy, enteropathy, X linked syndrome (IPEX) with intractable diarrhea. Ital J Pediatr. 2014;40:68.
11. Martin-Santiago A, Hervás JA, Hervás D, Rosell A, Caimari M, Carlos JC, et al. Diagnostic Value of the Skin Lesions in Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome. Pediatr Dermatol. 2013;30(6):e221-2.
12. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet. 2002;39:537-45.
13. Instituto Latino-Americano para Estudos da Sepse. Sepse: um problema de saúde pública. Brasília: CFM; 2015.
14. Kucuk ZY, Bleesing JJ, Marsh R, Zhang K, Davies S, Filipovich AH. A challenging undertaking: Stem cell transplantation for immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. J Allergy Clin Immunol. 2016;137(3):953-5.e4.
15. Passerini L, Sio FRS, Porteus MH, Bacchetta R. Gene/ Cell Therapy Approaches for Immune Dysregulation Polyendocrinopathy Enteropathy X Linked Syndrome. Curr Gene Ther. 2014;14(6):422-8.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888