Número Atual: Janeiro-Março 2017 - Volume 1 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Pedro Giavina-Bianchi
- L. Karla Arruda
- Marcelo V. Aun
- Regis A. Campos
- Herberto J. Chong-Neto
- Rosemeire N. Constantino-Silva
- Fátima F. Fernandes
- Maria F. Ferraro
- Mariana P. L. Ferriani
- Alfeu T. França
- Gustavo Fusaro
- Juliana F. B. Garcia
- Shirley Komninakis
- Luana S. M. Maia
- Eli Mansour
- Adriana S. Moreno
- Antonio A. Motta
- Joao Bosco Pesquero
- Nathalia Portilho
- Nelson A. Rosário
- Faradiba S. Serpa
- Dirceu Solé
- Eliana Toledo
- Solange O. R. Valle
- Camila Lopes Veronez
- Anete S. Grumach

ARTIGO ESPECIAL
Diretrizes brasileiras para o diagnóstico e tratamento do angioedema hereditário - 2017
Brazilian guidelines for the diagnosis and treatment of hereditary angioedema - 2017
Pedro Giavina-Bianchi1; L. Karla Arruda2; Marcelo V. Aun1; Regis A. Campos3; Herberto J. Chong-Neto4; Rosemeire N. Constantino-Silva5; Fátima F. Fernandes6; Maria F. Ferraro2; Mariana P. L. Ferriani2; Alfeu T. França7; Gustavo Fusaro8; Juliana F. B. Garcia1; Shirley Komninakis5; Luana S. M. Maia2; Eli Mansour9; Adriana S. Moreno2; Antonio A. Motta1; Joao Bosco Pesquero10; Nathalia Portilho1; Nelson A. Rosário4; Faradiba S. Serpa11; Dirceu Solé12; Eliana Toledo13; Solange O. R. Valle7; Camila Lopes Veronez10; Anete S. Grumach5
DOI: 10.5935/2526-5393.20170005
1. Disciplina de Imunologia Clínica e Alergia, Universidade de Sao Paulo (USP), Sao Paulo, SP
2. Faculdade de Medicina de Ribeirao Preto, Universidade de Sao Paulo (FMRP-USP), Ribeirao Preto, SP
3. Departamento de Medicina Interna e Apoio Diagnóstico, Faculdade de Medicina da Bahia (UFBA), Salvador, BA
4. Departamento de Pediatria, Universidade Federal do Paraná (UFPR), Curitiba, PR
5. Imunologia Clínica, Faculdade de Medicina do ABC (FMABC), Sao Paulo, SP
6. Hospital do Servidor Público Estadual "Francisco Morato Oliveira", Sao Paulo, SP
7. Divisao de Imunologia, Departamento de Medicina Interna, Universidade Federal do Rio de Janeiro (UFRJ), Rio de Janeiro, RJ
8. Divisao de Imunologia Clínica e Alergia, Departamento de Pediatria - Universidade Federal de Minas Gerais (UFMG), MG
9. Divisao de Alergia e Imunologia Clínica, Departamento de Clínica Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas (UNICAMP), Campinas, SP
10. Departamento de Biofísica, Universidade Federal de Sao Paulo (UNIFESP), Sao Paulo, SP
11. Faculdade de Ciências da Santa Casa de Misericórdia de Vitória, Vitória, ES
12. Divisao de Alergia, Imunologia Clínica e Reumatologia, Departamento de Pediatria, Universidade Federal de Sao Paulo (UNIFESP), Sao Paulo, SP
13. Divisao de Imunologia Clínica e Alergia, Faculdade de Medicina de Sao José do Rio Preto (FAMERP), Sao José do Rio Preto, SP
Submetido em: 10/01/2017
Aceito em: 15/02/2017
Conflitos de interesse: os autores Giavina-Bianchi P, Aun MV, Campos RA, Chong-Neto HJ, Ferriani MP, Garcia JFB, Mansour E, Toledo E, Valle SO e Grumach AS referem ter recebido apoio financeiro e/ou honorários da CSL Boehring e Shire. Os autores Arruda LK, Ferraro MF, Maia LS, Motta AA, Moreno AS e Serpa FS referem ter recebido apoio financeiro e/ou honorários da Shire. Os autores Constantino-Silva RN, Fernandes FF, França AT, Fusaro G, Komninakis S, Pesquero JB, Portilho N, Rosário NA, Solé D e Veronez CL negam conflitos de interesse.
RESUMO
O angioedema hereditário é uma doença autossômica dominante caracterizada por crises de edema com o envolvimento de múltiplos órgaos. A doença é desconhecida por muitos profissionais da área da saúde e, portanto, subdiagnosticada. Os pacientes que nao sao diagnosticados e tratados adequadamente têm uma mortalidade estimada de 25% a 40%, devido ao angioedema da laringe, resultando em asfixia. O angioedema de alças intestinais é outra manifestaçao importante e incapacitante, que pode ser a principal ou a única durante uma crise da doença. Neste cenário, um grupo de especialistas da Associaçao Brasileira de Alergia e Imunologia (ASBAI) e do Grupo Brasileiro de Estudos sobre Angioedema Hereditário (GEBRAEH) atualizou as diretrizes para o diagnóstico e terapia do angioedema hereditário.
Descritores: Angioedema, angioedema hereditário, diagnóstico, tratamento.
POR QUE ESTUDAR O ANGIOEDEMA HEREDITARIO?
Desde a publicaçao das primeiras Diretrizes Brasileiras para Angioedema Hereditário (AEH) em 2011, o conhecimento sobre a doença aumentou, houve maior e mais precoce identificaçao dos pacientes e, ainda, novos tratamentos foram disponibilizados em nosso país, proporcionando uma abordagem melhor do AEH1-6. Neste cenário, um grupo de especialistas da Associaçao Brasileira de Alergia e Imunologia (ASBAI) e do Grupo Brasileiro de Estudos sobre Angioedema Hereditário (GEBRAEH) atualizou as diretrizes para o diagnóstico e terapia do AEH.
O Prof. William Osler, que definiu a natureza hereditária do AEH, afirmava que "A medicina é uma ciência e uma arte". Embora o desenvolvimento e aplicaçao de diretrizes baseadas em evidências científicas seja fundamental para a prática da medicina enquanto ciência, auxiliar os pacientes com as suas características biopsicossociais de forma personalizada é uma forma de praticar a medicina como uma arte.
O AEH é ainda desconhecido e subdiagnosticado por muitos profissionais da área da saúde. O longo tempo transcorrido entre o início dos sintomas até o diagnóstico, bem como deste até o acesso à terapia, aumenta a morbidade relacionada à doença, afetando a qualidade de vida dos pacientes e de suas famílias1,2,5-9. Observou-se que pacientes de AEH consultaram, em média, 4,4 médicos antes de serem corretamente diagnosticados e que 65% deles tiveram erro de diagnóstico no passado10,11. Embora o AEH represente uma minoria de casos de angioedema e a estimativa inicial de sua prevalência tenha sido de 1:50.000 (variando de 1:10.000 a 1:150.000), novos subgrupos de pacientes têm sido descritos, tornando a doença mais comum do que se pensava anteriormente6,12-15. Os médicos, bem como outros profissionais da saúde, devem estar cientes sobre a apresentaçao clínica e exames laboratoriais de avaliaçao que podem sugerir o diagnóstico de AEH. Além disso, os especialistas em alergia e imunologia devem ser atualizados sobre os progressos em relaçao ao diagnóstico e tratamento dos pacientes.
Os pacientes que nao sao tratados adequadamente têm uma mortalidade estimada de 25% a 40%, devido ao angioedema da laringe, resultando em asfixia5,12,16,17. Nos Estados Unidos, o AEH é responsável por 15.000 a 30.000 consultas em setores de emergência por ano, que muitas vezes levam à hospitalizaçao e internaçao em unidades de terapia intensiva18,19. O angioedema de alças intestinais é outra manifestaçao importante e incapacitante do AEH, que pode ser a principal ou a única durante uma crise da doença. Os pacientes muitas vezes sao equivocadamente diagnosticados com abdômen agudo cirúrgico, passando por cirurgias desnecessárias12,20,21. Estima-se que os pacientes de AEH sofram algum grau de incapacidade durante 20 a 100 dias do ano4,9.
O QUE É ANGIOEDEMA HEREDITARIO?
Angioedema é um edema localizado, nao inflamatório, assimétrico, desfigurante e autolimitado da derme profunda, tecidos subcutâneos ou submucosa, decorrente da vasodilataçao e aumento da permeabilidade vascular. O termo AEH é aplicado para o angioedema recorrente causado por excesso de bradicinina cuja forma de herança é autossômica dominante.
QUAL É A CAUSA DO ANGIOEDEMA HEREDITARIO?
Como entidade clínica, Quincke descreveu pela primeira vez o AEH em 188222-24, enquanto Osler estabeleceu a sua natureza hereditária em 188825-29. A primeira alteraçao bioquímica associada à doença, a deficiência do inibidor de C1 (C1-INH), só foi identificada 75 anos mais tarde. Os pacientes com AEH apresentam um defeito quantitativo ou qualitativo do C1-INH, enzima da superfamília SERPINA que atua como serinoprotease30-32. Inicialmente, foi reconhecida por sua atividade na inibiçao do Sistema Complemento, tanto nas vias clássica como das lectinas, sem a qual resultaria em um sistema excessivamente ativado33. Subsequentemente, o C1-INH também foi reconhecido como um inibidor de várias proteases, incluindo a calicreína do plasma, fatores de coagulaçao XII (FXII) e XI e plasmina. Portanto, o C1-INH além de inibir o Sistema Complemento, participa na regulaçao dos sistemas de contato, coagulaçao e de fibrinólise34-36.
Episódios de angioedema foram inicialmente atribuídos a fatores formados durante a ativaçao do sistema do complemento, incluindo um fragmento de C2 (C2 cinina) associado com a vasodilataçao e o aumento da permeabilidade. Estudos adicionais revelaram que a deficiência de C1-INH resulta em excesso de ativaçao do sistema de contato (sistema calicreína-cinina) com o aumento da produçao de bradicinina (Figura 1). Este mediador se liga ao seu receptor B2, que é constitutivamente expresso em células endoteliais, e interfere nas junçoes endoteliais, aumentando a permeabilidade vascular e induzindo ao angioedema37,38. A bradicinina também estimula a produçao de óxido nítrico, o qual desencadeia vasodilataçao por contraçao do citoesqueleto39. As evidências científicas indicam que a bradicinina é o principal mediador do AEH40,41.
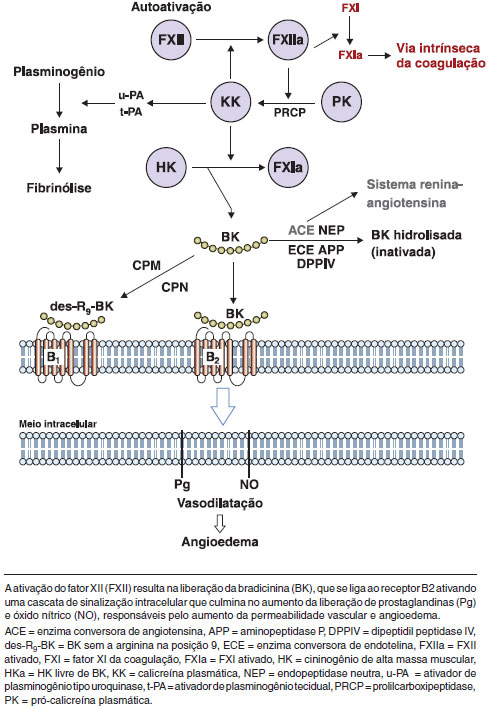
Figura 1 Ativaçao do sistema calicreína-cininas no angioedema hereditário
Nussberger et al. (1999) avaliaram amostras de plasma de pacientes com AEH e observaram que os níveis de bradicinina estavam mais elevados no sangue coletado no braço edemaciado, em comparaçao com aquele nao afetado, isto é, sem edema42. Um outro estudo mostrou uma diminuiçao da permeabilidade vascular em ratos duplamente knockout para o C1-INH e o receptor B2 de bradicinina (BDKRB2), demonstrando que a via de bradicinina/BDKRB2 desempenha um papel importante no angioedema43. O desenvolvimento de novos tratamentos, como o antagonista do receptor B2 de bradicinina e o inibidor de calicreína, reforçou o papel da bradicinina como principal mediador de AEH44,45.
Em conclusao, o AEH com deficiência de C1-INH ocorre devido à diminuiçao da quantidade desta enzima ou como consequência da produçao de uma enzima disfuncional. Entre os pacientes com AEH, os níveis funcionais de C1-INH sao, tipicamente, entre 5% e 30% do valor normal, apesar do nível funcional esperado de cerca de 50%, devido ao fato de que quase todos os pacientes sao heterozigotos para a mutaçao, possuindo um alelo funcional do gene C1-INH4,12. As razoes para a diminuiçao desproporcional da atividade funcional do C1-INH, com níveis abaixo do esperado, ainda nao estao claras, sugerindo a ocorrência de mecanismos adicionais no AEH.
Em 2000, Bork et al. identificaram um novo grupo de pacientes com AEH com C1-INH normal, nesta ocasiao denominado tipo III46. Subsequentemente, mutaçoes no gene codificador do fator de coagulaçao XII (FXII) foram descritas em parte das famílias de pacientes com AEH com C1-INH normal e este tipo de AEH foi designado como AEH-FXII47. O FXII tem um papel central nas fases iniciais de ativaçao do sistema de contato, aumentando a síntese de bradicinina48. Observaçoes iniciais sugeriram que as mutaçoes descritas no FXII levariam a ganho de funçao e a subsequente elevaçao da produçao de bradicinina. No entanto, estudos subsequentes nao confirmaram esta observaçao e o papel funcional das mutaçoes nao foi totalmente elucidado49,50. Estudo recente utilizando FXII recombinante, com e sem mutaçao, mostrou que mutaçoes do gene introduzem novos locais no FXII que sao sensíveis à clivagem enzimática pela plasmina, tornando o FXII anormalmente sensível à plasmina. Os FXII mutantes ativam-se rapidamente após clivagem pela plasmina, escapam da inibiçao de C1-INH e induzem formaçao excessiva de bradicinina. Curiosamente, os autores forneceram evidências de que os análogos de lisina, incluindo o ácido tranexâmico e o ácido épsilon-aminocapróico, podem atenuar este mecanismo, explicando o seu valor terapêutico em pacientes com AEH-FXII51. No entanto, nem todos os pacientes com AEH com C1-INH normal possuem mutaçao no FXII, apresentando o AEH de causa desconhecida (AEH-U)52. Como no AEH com deficiência de C1-INH, há evidências clínicas que sugerem que a bradicinina é também o mediador desencadeante das crises de AEH nos pacientes com C1-INH normal, que tem como principal característica a associaçao a situaçoes de hiperestrogenismo. Também se caracterizam por ausência de urticária, agravamento do quadro clínico com o uso de inibidores de enzima conversora da angiotensina (ECA), falta de resposta a anti-histamínicos e corticosteroides e por melhora quando do uso de bloqueador do receptor B2 da bradicinina4,53.
Como relatado, pacientes com AEH, especialmente aqueles com C1-INH normal, pioram dos sintomas com o uso de estrogênio exógeno, como ocorre durante a terapia de contracepçao e de reposiçao hormonal53. Os mecanismos por detrás dessa deterioraçao sao apenas parcialmente conhecidos. A regiao promotora do gene FXII contém um elemento de resposta ao estrogênio e foi demonstrado o aumento da transcriçao de mRNA de FXII em resposta ao hormônio54. É provável que o estrogênio também contribua para a regulaçao da expressao do receptor B2 da bradicinina, module a cascata de calicreína-cinina e reduza os níveis de C1-INH55,56.
QUAIS SAO OS TIPOS DE ANGIOEDEMA HEREDITARIO?
Em uma tentativa de padronizar a nomenclatura, um consenso internacional de especialistas em AEH propôs uma classificaçao para "Angioedema sem urticas", que se baseou principalmente na presença ou ausência de deficiência de C1-INH4. Foram definidos três tipos de AEH:
1. AEH com deficiência quantitativa de C1-INH (anteriormente designado como AEH C1-INH de Tipo I)
AEH caracterizado por diminuiçao quantitativa do C1-INH, com níveis inferiores a 50% dos valores normais, e consequente diminuiçao da atividade funcional. Este fenótipo é a forma mais prevalente de todos os casos de AEH (80-85% dos casos associados à deficiência de C1-INH)57.
2. AEH com disfunçao de C1-INH (anteriormente designado como AEH C1-INH de Tipo II)
AEH com níveis normais ou elevados de C1-INH, mas com comprometimento de sua funçao, por ser uma proteína anômala58.
3. AEH com C1-INH normal (anteriormente designado como AEH de Tipo III)
Identificada mais recentemente, esta forma de AEH afeta principalmente as mulheres, mas também foi identificada em indivíduos do sexo masculino, e é caracterizada por sintomatologia clínica semelhante ao AEH com deficiência de C1-INH, história familiar positiva e ausência de déficit de C1-INH15,53. AEH com C1-INH normal tem sido associado a maiores níveis séricos de estrogênio (gravidez e administraçao exógena) e a mutaçoes no gene que codifica o FXII em um subgrupo de pacientes (AEH-FXII). No entanto, numa percentagem significativa dos casos, o defeito genético é ainda desconhecido. Portanto, dois subtipos sao reconhecidos: AEH com C1-INH normal e mutaçao de FXII (AEH-FXII); e AEH com C1-INH normal e defeito genético desconhecido (AEH-U)1,2,5-9,52. Nao é recomendável usar mais o termo AEH tipo III, pois essa forma de AEH nao está associada à deficiência de C1-INH.
QUAIS SAO AS MANIFESTAÇOES CLINICAS TIPICAS DE ANGIOEDEMA HEREDITARIO?
A história clínica é um componente importante do diagnóstico do AEH e foi bem caracterizada nas coortes dos Profs. Agostoni, Cicardi e Bork2,3,5,11. Pacientes com AEH sofrem de episódios recorrentes de edema envolvendo a pele e a submucosa de vários órgaos. O AEH nao está associado à urticária e prurido, mas, às vezes, os pacientes referem sensaçao de queimaçao na regiao do edema. Os locais mais comumente afetados sao: face, extremidades, genitália, orofaringe, laringe e o sistema digestório. No entanto, manifestaçoes clínicas raras, como dor de cabeça intensa, retençao urinária e pancreatite aguda, também podem ocorrer59.
A frequência e a gravidade das crises variam entre os pacientes. Tem sido relatado que 5% dos indivíduos com AEH sao assintomáticos, ao passo que 25% desenvolvem sintomas esporádicos5,6,13,15. Estudo retrospectivo analisando 131.110 crises em 221 pacientes com AEH mostrou que o edema de laringe ocorreu em menos de 1% dos episódios, embora mais de 50% dos pacientes tivessem, pelo menos uma vez, experimentado anteriormente esta manifestaçao5.
Episódios de AEH que nao recebem tratamento específico duram geralmente de 48 a 72 horas e nao melhoram com anti-histamínicos, corticosteroides, ou epinefrina. Embora muitas das crises ocorram espontaneamente, foram identificados diversos fatores desencadeadores: trauma (mesmo que de pouca intensidade), estresse, infecçao, menstruaçao, gravidez, consumo de álcool, mudanças de temperatura extremas, exercício, uso de inibidores da ECA e uso de estrogênio (anticoncepcionais e terapia de reposiçao hormonal). Na adolescência, pode haver aumento substancial da atividade da doença, em particular, nas jovens do sexo feminino, devido aos ciclos menstruais e à utilizaçao de contraceptivos orais contendo estrogênio2,3,5,11.
Pode ocorrer eritema serpiginoso como manifestaçao prodrômica ao episódio de AEH em alguns pacientes, mas a presença concomitante de urticária pruriginosa favorece o diagnóstico de angioedema histaminérgico, tornando improvável o diagnóstico de AEH5. No entanto, alguns casos de AEH acompanhados por urticária têm sido relatados. Além disso, irritabilidade, fraqueza, náusea e "sensaçao da gripe" também sao relatadas como pródromos2,3,5.
A história familiar de angioedema auxilia no diagnóstico de AEH, mas pode estar ausente em até um quarto dos casos.
Embora as manifestaçoes clínicas do AEH com C1-INH normal sejam semelhantes às de outros tipos de AEH, o início dos sintomas ocorre geralmente mais tarde e o curso da doença tende a ser mais benigno. Foi relatado que ocasionalmente observa-se hematomas nos locais afetados pelo angioedema. No entanto, a característica mais marcante do AEH com C1-INH normal é a maior frequência no sexo feminino e sua associaçao com o uso de estrogênio52. Na Figura 2, sugerimos uma lista de sinais de alerta e um acrônimo para estimular a suspeita diagnóstica e promover a conscientizaçao sobre o AEH.
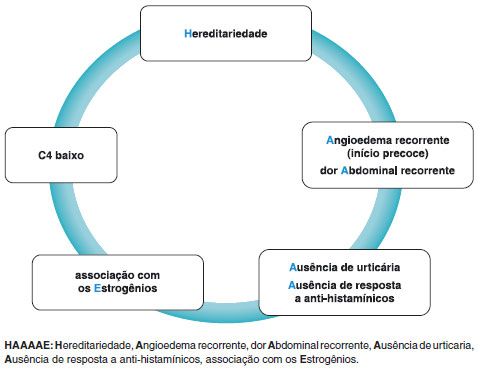
Figura 2 Sinais de alerta
COMO OS EXAMES LABORATORIAIS PODEM CONFIRMAR O DIAGNOSTICO?
Os indivíduos com suspeita clínica e/ou aqueles com história familiar de AEH devem ser investigados (Figura 3). Os níveis séricos de C4 podem ser utilizados como teste de triagem, tendo em vista que a deficiência quantitativa e/ou qualitativa de C1-INH conduz à ativaçao do Sistema de Complemento, resultando no consumo de C4, mesmo quando os pacientes estao fora do período de crise de angioedema. Em apenas 2-5% dos casos de AEH, os níveis de C4 se normalizam no período inter-crise5,10,16,17. Em contraste, a determinaçao dos níveis de C3 é desnecessária. Os níveis de C3 sao normais em pacientes com AEH, pois sua produçao é intensa e há outros fatores responsáveis pelo controle de seu consumo, como os fatores H e I da via alternativa do complemento.
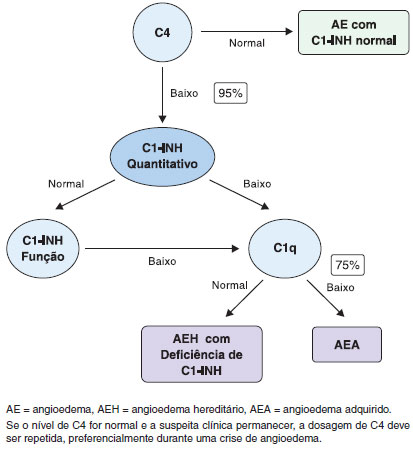
Figura 3 Algoritmo do diagnóstico do angioedema hereditário
Além da dosagem dos níveis séricos de C4, deve ser realizada a avaliaçao quantitativa e/ou funcional de C1-INH. Todos os profissionais da área da saúde envolvidos na assistência a pacientes com AEH devem assegurar que tais testes estejam disponíveis. Embora a determinaçao quantitativa de C1-INH seja um teste relativamente fácil de realizar, a avaliaçao da atividade funcional de C1-INH (teste qualitativo) deve ser realizada em laboratórios de referência12,14,19. Preferencialmente, os testes devem ser realizados imediatamente após a coleta da amostra para evitar a degradaçao dos fatores do complemento, com resultados falso-positivos. No entanto, como isso nao é viável na maioria dos casos, resultados confiáveis destes exames podem ser obtidos quando as amostras sao armazenadas corretamente e os testes realizados com metodologia adequada. Ao usar o ensaio cromogênico funcional, é crítico que as amostras sejam mantidas a -20 ºC em todos os passos do processo, incluindo o armazenamento e transporte, para a obtençao de resultados precisos60,61. Um conselho prático na avaliaçao funcional do C1-INH é evitar congelar e descongelar a mesma amostra mais de uma vez. A determinaçao da atividade funcional geralmente é realizada apenas quando a determinaçao quantitativa de C1-INH é normal (Figura 3). No entanto, alguns estudos sugerem que a atividade funcional poderia ser um teste de rastreio associado à determinaçao dos níveis de C4, considerando-se que a atividade funcional estaria reduzida em todos os pacientes com AEH por deficiência de C1-INH (AEH tipo I e II)61.
Se a suspeita clínica de AEH por deficiência de C1-INH permanecer na presença de níveis normais de C4, o teste deve ser realizado novamente durante uma crise angioedema, sempre que possível, pois, como mencionado anteriormente, os níveis de C4 podem estar normais entre as crises (2-5%)62. Se o resultado do teste for novamente normal e os níveis quantitativos e funcionais de C1-INH forem normais, na presença de história familiar, sugere-se um diagnóstico de AEH com C1-INH normal, no qual esses parâmetros bioquímicos sao normais18.
A análise de SERPING1, o gene codificador do C1-INH, pode ser realizada em casos de diagnóstico indefinido, ou para fins de investigaçao. Mutaçoes podem ser identificadas em uma das oito regioes adjacentes de exons ou exons/introns do gene, afetando a produçao da proteína e/ou a sua atividade enzimática. Nem todas as mutaçoes detectadas pelo teste genético de rotina sao, sem dúvida, causadoras de doença, e, às vezes, sao necessários testes genéticos de outros membros da família do paciente afetado que sejam isentos da doença. A avaliaçao molecular pode ser recomendada sempre que houver uma discrepância entre os testes de laboratório e o histórico clínico do paciente, em recém-nascidos ou crianças, e em situaçoes específicas, como pacientes sem história familiar ou com doença de início tardio, mas que apresentam sintomas clínicos sugestivos de AEH63,64.
No que se refere ao AEH com C1-INH normal, foram identificadas mutaçoes no gene do fator XII da coagulaçao (f12) em uma parcela dos pacientes. Inicialmente, o sequenciamento genético detectou duas mutaçoes missenses diferentes, localizadas no exon 9: uma mutaçao que conduz à substituiçao de treonina por lisina (p.Thr328Lys) e outra, de treonina por arginina (p.Thr328Arg)47. Subsequentemente, duas novas mutaçoes foram publicadas: a eliminaçao de 72 pares de bases (c.971_1018+24del72) em uma família originária da Turquia e uma duplicaçao de 18 pares de bases (c.892_909dup) descrita em pacientes da Hungria65,66.
No angioedema adquirido com deficiência de C1-INH (AEA), uma condiçao associada com doenças linfoproliferativas ou autoimunes, paraproteínas induzem à ativaçao e consumo dos componentes do complemento. Portanto, a determinaçao dos níveis de C1q, sendo este reduzido em 75% dos pacientes, pode ajudar a diferenciar o AEH do AEA. Além do consumo de C1q, anticorpos anti-C1-INH podem ser observados em AEA associada a doença autoimune67.
QUAIS SAO OS CRITÉRIOS DIAGNOSTICOS DO AEH?
Foram propostos critérios para padronizar o diagnóstico de AEH com deficiência de C1-INH (Tabela 1). De acordo com esses critérios, a presença de AEH é confirmada quando os pacientes atendem a um critério clínico primário e um critério bioquímico12,20,21. Cabe ressaltar que esses critérios nao sao absolutos e que o histórico clínico é preponderante, especialmente em localidades onde os exames laboratoriais nao estao disponíveis. Em casos selecionados, um teste terapêutico pode ajudar no estabelecimento do diagnóstico de AEH.

O diagnóstico de AEH com C1-INH normal é considerado em pacientes com angioedema recorrente nao associado a urticária, com atividade e níveis normais de C1-INH no plasma. Histórico de mais de um membro da família afetados e predomínio de pacientes do sexo feminino tornam o diagnóstico mais provável. Neste momento, o único teste de confirmaçao para AEH com C1-INH normal é o estudo do gene F12, buscando uma das mutaçoes descritas anteriormente (AEH-FXII)9,18.
O QUE NAO É ANGIOEDEMA HEREDITARIO?
Foram descritos dois mecanismos fisiopatológicos principais de angioedema: o angioedema induzido pela ativaçao de mastócitos e/ou basófilos, resultando na liberaçao de histamina e outros mediadores (angioedema histaminérgico), e o angioedema devido ao excesso de bradicinina (angioedema mediado pela bradicinina ou nao-histaminérgico), como visto no angioedema hereditário, angioedema adquirido com deficiência de C1-INH (AEA), e angioedema induzido por inibidores da ECA ou por outros medicamentos envolvidos no metabolismo de bradicinina4,26. Recentemente, uma classificaçao de angioedema por endótipos foi proposta, abordando a etiologia e/ou mecanismo fisiopatológico dos diferentes tipos desta entidade clínica (Figura 4)22,24,30-32.
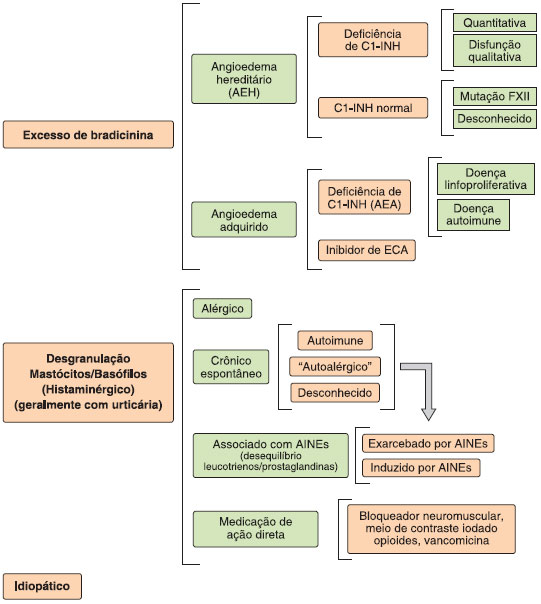
Figura 4 Classificaçao do angioedema por endótipos22,24
Os principais diagnósticos diferenciais do AEH sao os outros tipos de angioedema, principalmente aqueles com apresentaçao crônica ou recorrente. O tipo mais frequente de angioedema recorrente é o histaminérgico, que é normalmente associado a urticária e pode ser induzido ou exacerbado pela utilizaçao de drogas anti-inflamatórias nao-esteroidais (AINEs)25,27-29,33. Portanto, este angioedema apresenta algumas características que o diferenciam do AEH, incluindo a presença de urticária, melhora com anti-histamínicos e desencadeamento dos sintomas pelo uso de AINEs. No entanto, o angioedema histaminérgico crônico pode se apresentar sem urticária e os AINEs estao entre as principais causas de angioedema, mesmo naqueles pacientes que nao apresentam urticária22.
As diretrizes atuais para o tratamento de angioedema/urticária crônico espontâneo destacam o fato de que alguns pacientes nao responderao a doses convencionais de anti-histamínicos e podem necessitar de aumento de dose atingindo até quatro vezes as doses diárias habitualmente recomendadas para controlar os sintomas. Portanto, para confirmar ou descartar a natureza histaminérgica do angioedema, um teste terapêutico com anti-histamínicos, usando quatro vezes a dose recomendada por um período de tempo de aproximadamente 6 semanas, é suficiente para avaliar sua resposta ao tratamento. Foi demonstrada a segurança do aumento da dose de anti-histamínicos, incluindo cetirizina, levocetirizina, desloratadina, fexofenadina e rupatadina68.
Em relaçao às formas adquiridas de angioedema mediado por bradicinina, é muito importante perguntar ao paciente sobre o uso de inibidores da ECA. Como a ECA é a principal enzima envolvida na degradaçao da bradicinina, sua inibiçao leva ao aumento das concentraçoes séricas deste mediador e pode causar angioedema. O inibidor da ECA deve ser interrompido em todos os pacientes com angioedema recorrente, mesmo se o angioedema começou vários anos após o início do tratamento. Até 0,7% dos indivíduos que tomam inibidores da ECA apresentam angioedema recorrente, com aumento do risco entre os afrodescendentes, fumantes, idosos e no sexo feminino69,70. O angioedema induzido por ECA envolve mais frequentemente a face, língua, orofaringe e laringe; no entanto, foram relatados casos esporádicos de episódios abdominais. O tempo médio para o início dos sintomas de angioedema é de 1,8 anos, no entanto, os sintomas ocorrem no primeiro mês do uso da medicaçao em 25% dos casos e podem ocorrer até 10 anos após o início da droga71. Embora as crises de angioedema induzidas por ECA podem assemelhar-se àquelas de AEH, os pacientes apresentarao níveis normais de C4 e de C1q, e níveis quantitativos e/ou funcionais normais de C1-INH. Mais raramente, os bloqueadores dos receptores da angiotensina II (BRA) e as gliptinas também podem induzir angioedema72. As gliptinas sao agentes hipoglicêmicos orais que inibem a peptidase dipeptidil IV, outra enzima envolvida no catabolismo de bradicinina.
O denominado angioedema adquirido é uma outra forma adquirida de angioedema que está associado à deficiência de C1-INH (AEA), mas sem herança genética. No AEA, o aparecimento dos sintomas ocorre mais tardiamente, nao existe história familiar de angioedema e a doença deve-se ao consumo do C1-INH ou à produçao de autoanticorpos neutralizantes do C1-INH, associados com doenças linfoproliferativas ou doenças autoimunes, respectivamente12,73,74. Como consequência, a atividade do C1-INH é baixa, o sistema do complemento está ativado e o C1q geralmente reduzido, uma característica particular que pode ajudar no diagnóstico diferencial75,76. Além da funçao do C1-INH abaixo de 50% do normal, os níveis de antígenos de C1-INH sao geralmente reduzidos, embora a presença do C1-INH clivado possa resultar em níveis antigênicos de C1-INH normais em cerca de 20% dos pacientes75,77. Como há grande sobreposiçao dos AEA com deficiência de C1-INH associados a autoanticorpos e às doenças linfoproliferativas, sugere-se sua classificaçao como uma mesma doença4.
O angioedema nao histaminérgico idiopático é caracterizado quando nao-hereditário, todas as causas conhecidas de angioedema foram excluídas e os sintomas persistem apesar do tratamento com altas doses de anti-histamínicos, normalmente até 4 vezes a dose padrao de segunda geraçao, nao-sedativo anti-histamínicos, como já referido. Há certa evidência de que a bradicinina possa ser o mediador envolvido no angioedema nao-histaminérgico idiopático. No entanto, a evidência nao é definitiva, tendo em conta que em alguns pacientes outros mediadores vasoativos, incluindo cisteinil-leucotrienos, prostaglandinas e o fator ativador-plaquetas possam desempenhar um papel. O tratamento de pacientes com angioedema nao-histaminérgico idiopático é, muitas vezes, um desafio na prática clínica e nao há recomendaçoes definitivas para o tratamento. A eficácia do ácido tranexâmico e icatibanto em aliviar os sintomas tem sido relatada em casos individuais ou em poucos casos. A ciclosporina e o omalizumabe também têm sido utilizados, indicando a heterogeneidade deste grupo de pacientes. Devemos especular que esta classificaçao pode incluir pacientes com AEH com C1-INH normal, sem histórico familiar, e também pacientes com urticária/angioedema crônicos e resistentes a anti-histamínicos, pois, até 10% desses pacientes apresentam angioedema recorrente sem urticária4,24,68.
COMO TRATAR OS PACIENTES COM ANGIOEDEMA HEREDITARIO?
A educaçao e a orientaçao sao as açoes iniciais mais importantes para evitar consequências graves do AEH e para melhorar a qualidade de vida dos pacientes e de seus familiares. Os pacientes devem receber informaçoes por escrito que sejam relevantes sobre o AEH, incluindo medidas preventivas e um plano de açao para tratamento de crise.
A identificaçao e a eliminaçao de fatores desencadeantes, como o estresse e o trauma, podem reduzir o risco de crises. Esportes de alto impacto e passatempos que tenham risco de trauma sao contraindicados, da mesma forma que medicamentos que possam induzir ou prolongar uma crise de AEH, tais como inibidores da ECA, bloqueadores de receptores de Angiotensina II (BRA), medicamentos contendo estrogênio e gliptinas. Pacientes que necessitam de contracepçao devem receber apenas progestágenos. Recomenda-se a vacinaçao contra hepatites A e B, pois, produtos derivados do sangue podem ser utilizados no tratamento do AEH, embora nao se registre infecçao por estes vírus em pacientes que utilizaram os medicamentos atualmente disponíveis.
Todos os parentes de primeiro grau de pacientes com AEH devem ser rastreados, e os pacientes devem receber aconselhamento genético. Um estudo mostrou que apenas a metade das famílias dos pacientes com AEH havia sido testada11.
Como o AEH está associado à morbidade e mortalidade significativas, uma estratégia que envolva a prevençao cuidadosa e o tratamento das crises é essencial na abordagem apropriada do paciente. A experiência histórica em grandes centros de países desenvolvidos tem demonstrado que 25-40% dos pacientes podem desenvolver asfixia e morrer se nao forem tratados adequadamente5,16,78,79.
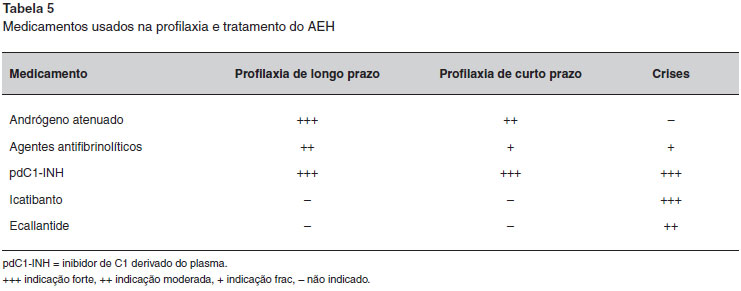
A farmacoterapia do AEH é dividida em três modalidades: profilaxia em longo prazo, profilaxia em curto prazo e tratamento das crises.
COMO A PROFILAXIA DE LONGO PRAZO DEVE SER REALIZADA?
O objetivo da profilaxia em longo prazo no AEH é reduzir a frequência e a gravidade das crises. Essas duas variáveis podem diferir muito e enquanto alguns pacientes podem ser assintomáticos, outros experimentam crises duas vezes por semana, com sintomas praticamente contínuos. Há evidência científica demonstrando a eficácia da profilaxia de longo prazo, mas também há preocupaçao com os efeitos adversos associados aos medicamentos4,6,80-82. Portanto, a primeira pergunta a respeito da profilaxia de longo prazo deve ser se os pacientes realmente necessitam deste tipo de tratamento.
Em geral, os indivíduos com sintomas frequentes ou histórico de crises de angioedema envolvendo as vias aéreas superiores devem ser tratados profilaticamente83. No entanto, nao há consenso sobre a frequência mínima e a gravidade dos episódios que justificam a profilaxia de longo prazo, e o impacto das crises de angioedema na qualidade de vida do paciente é um aspecto decisivo9,84. Os pacientes sofrem incapacitaçao física e emocional substancial, durante e entre as crises, muitas vezes impossibilitando as atividades diárias. Frequentemente desenvolvem ansiedade e/ou depressao85.
A introduçao de terapias novas e eficazes para as crises de AEH mudou a indicaçao da terapia preventiva, pois o tratamento somente durante os ataques poderia evitar os efeitos adversos decorrentes dos medicamentos profiláticos contínuos4,6. Portanto, principalmente em nosso país, outra variável importante a ser considerada é se o paciente tem acesso aos medicamentos apropriados para o tratamento da crise de AEH58. Os especialistas concordam que os pacientes que apresentam mais de uma crise grave por mês, apesar de serem tratados adequadamente durante as crises, devem receber profilaxia de longo prazo12,14. Em resumo, o tratamento deve ser sempre personalizado e a indicaçao de profilaxia em longo prazo deve ser baseada nas quatro variáveis citadas (Tabela 2).

Três modalidades de tratamento para a profilaxia em longo prazo têm sido utilizadas ao redor do Mundo (Tabela 3): andrógenos atenuados, agentes antifibrinolíticos e concentrado de C1-INH derivado de plasma (pdC1-INH). As duas primeiras estao disponíveis há muito tempo, têm custos mais baixos e nao necessitam de acesso intravenoso para administraçao. No entanto, existem preocupaçoes sobre os seus efeitos colaterais, que podem ser evitados com a terapia de reposiçao utilizando o pdC1-INH4,6. Há que se ressaltar que o uso intravenoso do C1-INH derivado de plasma com o auxílio de "portocath", provavelmente, foi associado a quadros de trombose venosa. Mais recentemente, foi publicado o uso de pdC1-INH por via subcutânea em profilaxia do AEH86.
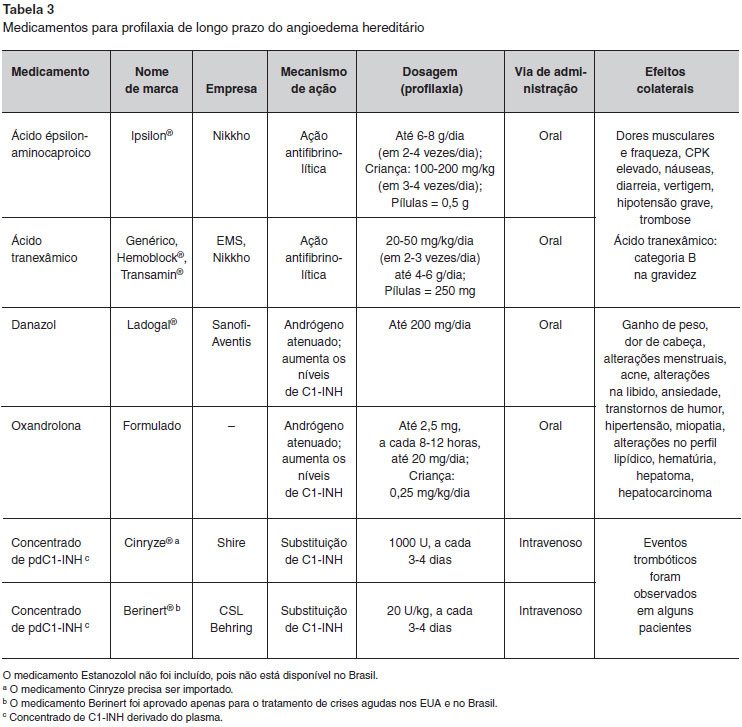
Os agentes antifibrinolíticos (ácido épsilon-aminocapróico e ácido tranexâmico) sao eficazes na prevençao de crises de AEH em um terço dos pacientes87-88. Estes agentes antagonizam o sistema fibrinolítico, bloqueando a formaçao de plasmina e inibindo a atividade proteolítica de ativadores de plasminogênio e, consequentemente, a dissoluçao do coágulo. No entanto, o mecanismo pelo qual os agentes antifibrinolíticos previnem as crises de AEH permanece desconhecido e nao se observa aumento dos níveis séricos de C1-INH ou C4 nos pacientes sob esta terapia. A principal preocupaçao com o uso de agentes antifibrinolíticos é o risco de trombose, embora esta reaçao adversa nao tenha sido relatada89,90. Pacientes que iniciarem a profilaxia de longo prazo com agentes antifibrinolíticos devem realizar testes de funçao hepática e devem ser avaliados em termos de trombofilia caso tenham histórico pessoal ou familiar de doença tromboembólica91,92. O ácido tranexâmico é mais potente do que o ácido épsilon-aminocapróico e tem uma incidência menor de efeitos adversos. O início do efeito terapêutico dos agentes antifibrinolíticos é de aproximadamente 48 horas após sua administraçao93-95.
Durante anos, a profilaxia de longo prazo do AEH mais eficaz era o uso de andrógenos atenuados (danazol, estanazolol e oxandrolona), que resultam em aumento nos níveis de C1-INH e C4 e reduzem a frequência das crises1,4,6. Andrógenos anabolizantes sao mais eficazes que agentes antifibrinolíticos no controle de AEH e, de forma geral, sao o tratamento de escolha, a menos que haja alguma contraindicaçao, como gravidez, infância e reaçoes adversas1,4,6. No Brasil, o andrógeno atenuado mais utilizado é o danazol, que está disponível por meio do Programa de Medicamentos de Alto Custo do governo.
Protocolos anteriores sugeriam a dose máxima de 600 mg/dia de danazol, iniciando com dose menor de 200 mg/dia e aumentando se necessário (Protocolo de Budapeste), ou começando com dose mais elevada de 600 mg/dia e reduzindo a medida do possível (Protocolo Italiano)1. Posteriormente, devido à preocupaçao com as reaçoes adversas, o Consenso de Gargnano (2012) definiu a dosagem máxima de danazol em 200 mg/dia96. Em casos graves, nos quais a dose máxima de andrógeno atenuado nao é suficiente para prevenir as crises, agentes antifibrinolíticos podem ser utilizados em combinaçao97.
As reaçoes adversas aos andrógenos atenuados sao relacionadas à dose, e incluem hepatotoxicidade e virilizaçao. Outras reaçoes adversas incluem ganho de peso, dor de cabeça, alteraçoes menstruais, acne, alteraçoes na libido, ansiedade, perturbaçoes do humor, hipertensao, miopatia, mudanças no perfil lipídico e hematúria. Embora existam evidências de alteraçoes do perfil lipídico dos pacientes, a associaçao entre danazol e aterosclerose é controversa98-100. Estes efeitos adversos tendem a desaparecer após a descontinuaçao do medicamento. Em indivíduos que recebem andrógenos, o perfil lipídico e os níveis de enzimas hepáticas devem ser avaliados a cada seis meses. Se houver desenvolvimento de dislipidemia ou lesao hepática, a droga deve ter sua dose reduzida ou ser descontinuada até a melhora do paciente101-104. Como foram relatados casos de adenomas hepáticos e carcinoma hepatocelular, ultrassonografia do fígado deve também ser realizada a cada 12 meses. O desenvolvimento de tumores do fígado em pacientes que estao utilizando o danazol tem sido associado à utilizaçao de doses mais elevadas (400-800 mg), duraçao mais longa do tratamento e falta de controle de lesao de fígado4,105. Embora muitos pacientes se queixem de efeitos adversos induzidos por andrógenos atenuados, a maioria dos pacientes com AEH pode se beneficiar, pelo menos moderadamente, do uso de andrógeno, e o perfil de risco é aceitável1.
Na profilaxia de longo prazo, o ajuste da dose dos medicamentos baseia-se nos sintomas clínicos do paciente ao invés de parâmetros bioquímicos, tendo em vista que o benefício clínico é geralmente obtido em doses mais baixas que as exigidas para alterar significativamente os níveis dos componentes do complemento1. A profilaxia de longo prazo do AEH com andrógenos atenuados ou agentes antifibrinolíticos é satisfatória para a maioria dos pacientes, mas tem a desvantagem do uso de medicaçao diária. Em alguns pacientes, é impraticável devido a reaçoes adversas, principalmente em mulheres, ou devido à falta de resposta1. Acrescenta-se, ainda, a nao recomendaçao para gestantes e crianças.
Os concentrados de pdC1-INH têm sido utilizados recentemente para profilaxia de longo prazo do AEH, e têm demonstrado serem seguros e eficazes82,106-110. Os produtos disponíveis sofrem nanofiltragem e sao tratados com calor e detergente para evitar a transmissao de micróbios. Administrados por via intravenosa em intervalos regulares, a cada três a quatro dias, os concentrados de pdC1-INH constituem uma alternativa terapêutica adequada para indivíduos nos quais outras terapias nao podem ser usadas ou sao ineficazes. Poucos pacientes com eventos trombóticos associados ao pdC1-INH foram identificados pelo programa de controle de efeitos adversos do FDA e estao sob investigaçao111. Na avaliaçao da relaçao risco/benefício da profilaxia de longo prazo, enquanto há menos efeitos adversos com o pdC1-INH, favorecendo-o em comparaçao com os andrógenos atenuados e os agentes antifibrinolíticos, seu custo é maior e há necessidade de acesso venoso. Os indivíduos que têm reduçao significativa na qualidade de vida ou têm episódios graves e frequentes, e falham ou sao intolerantes aos andrógenos devem ser considerados para profilaxia de longo prazo com pdC1-INH108,112. Dois produtos mais recentes foram avaliados para profilaxia de longo prazo do AEH: um C1-INH recombinante (Ruconestr, que foi aprovado pelo FDA em 2014) e um concentrado de pdC1-INH para uso subcutâneo, este último supera a inconveniência do acesso venoso. Estudos têm demonstrado a eficácia e segurança de ambos113,114.
Em relaçao ao AEH com C1-INH normal, a experiência é restrita a relatos de caso. O ácido tranexâmico reduz a frequência e a gravidade dos episódios e pode ser considerado uma escolha adequada, com evidências sugerindo uma melhor resposta nestes pacientes em comparaçao aos pacientes de AEH com deficiência de C1-INH89,115. Além disso, a utilizaçao de progestágenos tem sido recomendada, com estudos mostrando sua eficácia52,64.
A melhor estratégia para a profilaxia de longo prazo do AEH deve ser escolhida dependendo do estado clínico e da qualidade de vida do paciente. Portanto, há de se considerar se é mais importante controlar as crises de AEH o mais rápido possível ou minimizar os potenciais efeitos adversos dos medicamentos. Independentemente da escolha, a eficácia de cada medicamento depende da adesao ao tratamento, que deve ser encorajada e avaliada.
COMO A PROFILAXIA DE CURTO PRAZO DEVE SER REALIZADA?
A profilaxia de curto prazo é indicada para pacientes submetidos a procedimentos médicos ou cirúrgicos que envolvam principalmente a regiao cervicofacial, com risco de angioedema das vias aéreas superiores, como tratamento dentário mais invasivo (extraçao dentária), amigdalectomia, cirurgia facial, endoscopia, broncoscopia e procedimentos cirúrgicos que exigem a intubaçao traqueal1,116-119. A decisao sobre a profilaxia de curto prazo deve ser tomada tendo em conta dois fatores: o risco associado ao procedimento a ser feito e a disponibilidade de tratamento para a crise aguda de AEH (Figura 5).
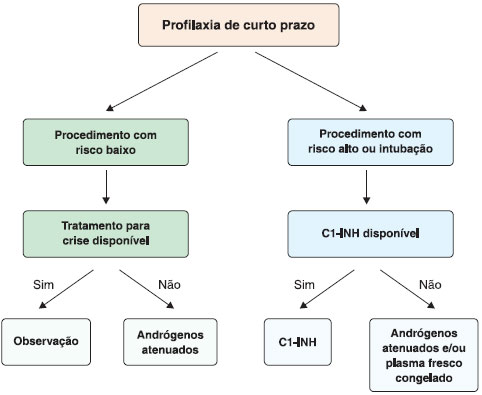
Figura 5 Profilaxia de curto prazo do angioedema hereditário com deficiência de C1-INH
Os agentes mais utilizados com sucesso incluem os concentrados de pdC1-INH, andrógenos atenuados e plasma fresco congelado. Como nao existem estudos comparativos avaliando os diferentes medicamentos para a profilaxia de curto prazo, as recomendaçoes atuais sao baseadas na opiniao de especialistas e em estudos de observaçao pequenos e nao controlados96.
O concentrado do pdC1-INH é o agente de escolha para a profilaxia de curto prazo, sendo seguro e eficaz em adultos, crianças e mulheres grávidas. Tem sido usado antes do procedimento em dose de 20 U/kg ou 500-1000 U, dependendo do fabricante62,120,121. Segundo dados relatados por Bork et al., doses de 500 UI reduzem as ocorrências de crises para 15%, doses de 1000 UI reduzem o desencadeamento para 10% e 1500 UI ainda sao acompanhados de sintomas em 5% das situaçoes117. Alguns autores consideram a administraçao de plasma fresco congelado (PFC) para a profilaxia de curto prazo, quando o C1-INH nao está disponível, sugerindo-se uma dose de 10 mL/kg (2-4 unidades para um adulto), cerca de uma a seis horas antes do procedimento122. Outra alternativa é a utilizaçao de danazol a uma dose de 10 mg/kg/dia, com uma dose máxima de 600 mg/dia, 200 mg três vezes por dia, durante 5-7 dias antes do procedimento e dois dias depois123. Os agentes antifibrinolítico, como o ácido tranexâmico, também têm sido utilizados nas doses de 25 mg/kg/dia, divididas em 2-3 vezes, até um máximo de 3-6 g/dia, 5 dias antes e 2-5 dias após o procedimento121. No entanto, sua eficácia é desconhecida e este agente só deve ser usado se outras opçoes nao estiverem disponíveis. Existem dados escassos referentes à profilaxia de curto prazo com rhC1-INH, Ecallantide, ou Icatibanto, mas especula-se que o rhC1-INH seja eficaz.
Mesmo com a realizaçao da profilaxia de curto prazo, os pacientes devem permanecer sob observaçao por 36 horas e devem ter fácil acesso à medicaçao de resgate, uma vez que o risco de angioedema após estes procedimentos nao pode ser completamente eliminado. Em algumas situaçoes, quando se espera que o risco do procedimento a ser realizado seja mínimo, e o acesso rápido aos agentes de terapia da crise está garantido, pode-se considerar a omissao da profilaxia de curto prazo. Nestes casos, deve-se instituir o tratamento da crise de AEH, caso esta ocorra, ao menor sinal de seu início6.
COMO AS CRISES DE ANGIOEDEMA HEREDITARIO DEVEM SER TRATADAS?
O tratamento das crises de AEH deve ser feito de acordo com sua gravidade (Tabela 4). Crises graves e/ou crises que envolvem o trato respiratório requerem tratamento urgente a fim de evitar potencial morbidade e mortalidade. Assim, pacientes com AEH devem ter acesso ao tratamento "sob demanda" para crises, sendo recomendável que tenham pelo menos duas terapias para usar em seu domicílio em casos de eventuais crises6,124,125. Independentemente da disponibilidade de todas as drogas usadas para tratar a crise de AEH, a prioridade deve ser sempre a manutençao da permeabilidade das vias aéreas, e os médicos nao podem perder o "momento certo" de intubaçao, caso esta seja indicada.
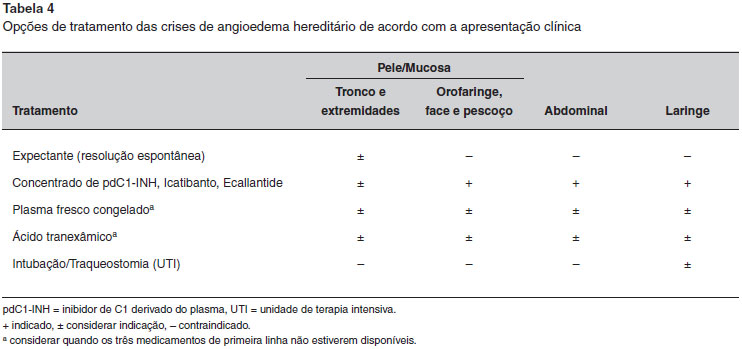
O objetivo da terapia da crise de AEH é inibir a síntese de bradicinina e a sua açao sobre as células endoteliais. Existem três medicamentos para tratar as crises de AEH: concentrado do pdC1-INH, Icatibanto e Ecallantide. Os dois primeiros estao disponíveis no Brasil. O plasma fresco congelado, que contém o C1-INH, pode ser utilizado quando estes três medicamentos de primeira linha nao estiverem disponíveis. Nenhum estudo controlado demonstrou sua eficácia e, embora o plasma fresco congelado possa controlar a maioria das crises de AEH, uma piora paradoxal pode ocorrer em alguns casos, pois o plasma também fornece cininogênio e pré-calicreína de alto peso molecular (HMW), que podem gerar mais bradicinina. Além disso, há riscos de transmissao de patógenos transmitidos pelo sangue e derivados, assim como de reaçoes transfusionais126,127. Assim, o plasma nao deve ser utilizado para o tratamento das crises de AEH, a menos que nenhuma outra opçao de tratamento de primeira linha esteja acessível4,6,14,128. Deve-se destacar que, ao contrário da anafilaxia e angioedema associados à desgranulaçao de mastócitos e basófilos, o angioedema mediado por bradicinina nao responde à administraçao de anti-histamínicos, glicocorticoides, ou epinefrina.
Andrógenos e agentes antifibrinolíticos sao inadequados para controlar as crises de AEH devido a demora para seu início de açao, sendo ineficazes na maioria das vezes. No entanto, tem-se observado que, nas fases iniciais de crises leves, estes medicamentos podem ser eficazes em atenuar os sintomas e prevenir a progressao para uma crise mais grave93,94,129.
Crises abdominais sao extremamente dolorosas e podem ser acompanhadas por vômitos, diarreia, ou ambos. Quando os pacientes apresentam crises abdominais graves, indica-se, além da terapia específica, tratamento sintomático com administraçao de fluidos, antieméticos e analgésicos. Podem ser necessários antiespasmódicos e narcóticos para tratar a dor intensa21.
A disfonia e a disfagia sao indicadores de progressao para uma crise grave de laringe. Geralmente, tais episódios desenvolvem-se lentamente ao longo de cerca de 8 horas, em média, e as alteraçoes da deglutiçao e de voz frequentemente precedem a obstruçao da laringe. No entanto, há relatos de edema de laringe de início rápido e os médicos devem ter isso em mente na avaliaçao dos pacientes. Os casos mais graves exigem a intubaçao imediata. Em tais casos, indica-se a terapia de oxigênio e o monitoramento por oximetria de pulso. Durante a laringoscopia e intubaçao, deve ser avaliada a necessidade de traqueostomia. Em casos de edema de laringe, pode ser prudente realizar a intubaçao profilática como uma medida inicial para manter a permeabilidade das vias aéreas e evitar a traqueostomia130.
REPOSIÇAO DE C1-INH
Os concentrados de C1-INH para uso intravenoso demonstraram ser eficazes e seguros no tratamento de todas as formas de crises de AEH por deficiência do C1-INH62,131-133. O pdC1-INH atua em várias etapas dos quatro sistemas que funcionam de maneira integrada: de contato, coagulaçao, fibrinolítico e complemento. Há inibiçao do fator Xlla e da calicreína, com reduçao da produçao de bradicinina34,35. Dois concentrados do pdC1-INH, que passam por nanofiltragem e sao tratados com calor e detergente para evitar a transmissao de micróbios, estao atualmente disponíveis em todo o mundo: Berinertr e Cinryzer.
O Berinertr é um concentrado do C1-INH pasteurizado e nanofiltrado. O ensaio clínico "International Multicenter Prospective Angioedema C1-INH Trial 1 - IMPACT1" foi um estudo internacional, prospectivo, multicêntrico, controlado por placebo, duplo-cego e randomizado que avaliou a eficácia do Berinertr em 125 pacientes com AEH por deficiência de C1-INH, demonstrando que este concentrado administrado por via intravenosa a uma dose de 20 U/kg é eficaz para tratamento das crises abdominais e faciais132. O IMPACT2 foi um estudo aberto desenvolvido como uma extensao do IMPACT1 para avaliar a eficácia e segurança do tratamento com 20 U/kg de C1-INH para crises sucessivas de AEH. O estudo incluiu 57 pacientes, que foram acompanhados durante 24 meses, e mostrou que, em média, o início do alívio dos sintomas após o tratamento foi de 0,46 horas e a resoluçao completa em 15,5 horas. O estudo concluiu que uma única dose de pdC1-INH de 20 U/kg é segura e eficaz no tratamento de crises sucessivas de AEH em qualquer local do corpo. Além disso, nenhum anticorpo de bloqueio anti-C1-INH foi detectado nos pacientes131. Esta dose deve ser utilizada em crises de vias aéreas superiores e de face, que apresentam risco de vida em potencial. De acordo com estudos anteriores, uma dose de 500 ou 1000 U pode ser suficiente em crises de menor gravidade, como o angioedema de extremidades e nas crises abdominais62,134,135. Berinertr está atualmente licenciado na Europa, Estados Unidos e no Brasil, e é aprovado para autoadministraçao domiciliar. O fármaco é geralmente bem tolerado e nao há evidência de doença tromboembólica ou de transmissao de agentes infecciosos associados com a sua administraçao136,137.
Cinryzer é um concentrado de pdC1-INH nanofiltrado aprovado para o tratamento e a prevençao de crises de AEH na Europa, apenas para a prevençao nos Estados Unidos e ainda nao aprovado no Brasil. Estudos, incluindo dois estudos duplo-cegos, randomizados, controlados por placebo, demonstraram sua eficácia. Da mesma forma que com o Berinertr, a autoadministraçao de Cinryzer reduziu significativamente a duraçao e a gravidade das crises, além da necessidade de analgésicos. Uma dose de 1000 U é utilizada para o tratamento das crises, a qual pode ser repetida uma hora mais tarde, se necessário109,138.
Ruconestr, um C1-INH recombinante humano (rhC1-INH) obtido a partir do leite de coelhas transgênicas, foi desenvolvido para o tratamento das crises de AEH. Dois estudos clínicos duplo-cegos, randomizados, controlados com placebo diferentes foram realizados na Europa e na América do Norte e demonstraram a eficácia e segurança do rhC1-INH, sem eventos adversos trombóticos observados139,140. Devido a sua glicosilaçao original, sua meia-vida é mais curta que a dos C1-INH derivados do plasma humano, e espera-se que o rhC1-INH seja mais eficaz no tratamento das crises de AEH que na profilaxia, mas um estudo recente demonstrou benefícios na profilaxia de longo prazo114. Como reaçoes anafiláticas em pacientes sensibilizados por coelho sao raramente observadas, os pacientes devem ser indagados em relaçao a contato e reaçao alérgica anterior a coelho. A dose recomendada é de uma injeçao intravenosa de 50 U/kg para adultos com um peso corporal inferior a 84 kg e uma dose de 4200 U (dois frascos) para adultos com peso superior140. Ruconestr foi aprovado na Europa e nos EUA, mas nao no Brasil. O rhC1-INH beneficiará especialmente aqueles pacientes que nao desejam ser tratados com um produto derivado de sangue humano por motivos religiosos, morais ou outros141.
ICATIBANTO
O Icatibanto (Firazyrr) é uma molécula sintética semelhante à bradicinina para injeçao subcutânea em pacientes com AEH com deficiência de C1-INH, agindo como um antagonista competitivo e seletivo do receptor B2 da bradicinina. A segurança e eficácia do Icatibanto foram avaliadas e demonstradas em três ensaios clínicos de fase III, multicêntricos, controlados, duplo-cegos e randomizados (Os estudos FAST - For Angioedema Subcutaneous Treatment). O tempo médio para o alívio significativo dos sintomas é menor com o Icatibanto do que com o ácido tranexâmico ou placebo142,143. Crises de AEH sao resolvidas mais rapidamente após o tratamento precoce com Icatibanto em comparaçao com o tratamento tardio; recomenda-se que o medicamento seja administrado nas primeiras seis horas após o início da crise144. Como ocorre com o Berinertr, o Firazyrr também é licenciado para autoadministraçao. O uso domiciliar é seguro e preferido pela maioria dos pacientes, que apenas relatam eritema e dor no local da injeçao que se resolvem espontaneamente145. O Icatibanto é licenciado para uso, inclusive para a autoadministraçao, a uma dose de 30 mg por via subcutânea, na Europa, Estados Unidos e Brasil. Se a resposta for inadequada ou houver recidiva dos sintomas, podem ser administradas injeçoes adicionais de 30 mg, em intervalos de 6 horas, até o máximo de três injeçoes em 24 horas. Atualmente, o Icatibanto está licenciado para os pacientes acima de 18 anos de idade. No entanto, estao em curso estudos abordando a sua utilizaçao em crianças146.
ECALLANTIDE
Ecallantide (Kalbitorr) é um potente inibidor da calicreína, aprovado para utilizaçao nos EUA, que acarreta a diminuiçao da produçao de bradicinina147. Dois grandes ensaios clínicos de fase III ("Evaluation of Dx-88's Effect in Mitigating Angioedema - EDEMA"), controlados com placebo, demonstraram a eficácia da medicaçao. A dose recomendada é de 30 mg administrada por via subcutânea, e o medicamento nao está aprovado para autoadministraçao domiciliar, porque anafilaxia foi observada em aproximadamente 3% dos doentes tratados148,149. Nao está disponível em nosso país.
CONCLUSAO
Todos os medicamentos mencionados para tratamento das crises de AEH sao altamente eficazes e nao existem estudos comparativos entre eles. Os concentrados do pdC1-INH e o C1-INH recombinante devem ser administrados por via intravenosa, requerendo um tempo ligeiramente maior para sua administraçao. O Icatibanto é administrado por via subcutânea e tem um início de açao mais rápido. No entanto, em cerca de 10% das crises tratadas, é necessária uma segunda injeçao44. Por outro lado, a taxa de recidivas das crises é muito baixa com os concentrados do pdC1-INH. Concentrados do pdC1-INH e o Icatibanto sao licenciados para autoadministraçao caseira. A Tabela 5 resume os medicamentos utilizados no tratamento e profilaxia do AEH.
COMO DEVE SER A ABORDAGEM DO ANGIOEDEMA HEREDITARIO EM SITUAÇOES ESPECIAIS?
Crianças e adolescentes
Na maioria dos pacientes, o início das crises do AEH com deficiência do C1-INH ocorre na primeira ou segunda década de vida, em média entre 4 e 12,5 anos e com apresentaçao variável5,150-154. O diagnóstico clínico e laboratorial nessa faixa etária é semelhante ao do adulto. Os sintomas sao raros durante a infância e, muitas vezes, se agravam na puberdade, principalmente em mulheres, estando associados aos níveis crescentes de estrogênio endógeno150,155. O início precoce e a frequência de episódios preveem a gravidade da doença na idade adulta5,154. Em relaçao ao AEH com C1-INH normal, sua incidência é extremamente rara em pacientes pediátricos155.
Embora a morte por asfixia possa ocorrer em qualquer idade, casos fatais devido ao edema de laringe sao menos comuns em pacientes pediátricos. No Reino Unido, 6,3% das crianças com AEH tiveram episódios com risco de vida reportados pelos pediatras156. Em uma série de 70 mortes, três ocorreram em pacientes com menos de 21 anos de idade130. O diagnóstico diferencial inclui alergia alimentar, crupe, pseudo crupe, aspiraçao de corpo estranho e epiglotite aguda. Por outro lado, crises abdominais sao frequentes em pacientes pediátricos, mas muitas vezes nao sao reconhecidas e o diagnóstico diferencial é amplo, incluindo apendicite aguda, linfadenite mesentérica, intussuscepçao, má rotaçao parcial com torçao intestinal, divertículo de Meckel, ovários policísticos, torçao testicular ou de ovário, hemorragia ou infarto intestinal, porfiria, e peritonite recorrente da febre familiar do Mediterrâneo. A resposta clínica e ultrassonográfica à terapia específica ajudam a diferenciar o episódio de AEH de outras doenças abdominais155.
Todas as crianças com suspeita clínica ou que sao parentes de primeiro grau de pacientes com AEH devem ser rastreadas. Antes da idade de 1 ano, os níveis de C4 e de C1-INH sao fisiologicamente mais baixos do que em adultos, e os testes devem ser repetidos após este período para se confirmar o diagnóstico de AEH com deficiência de C1-INH157-159). Os testes genéticos com o sangue periférico ou do cordao umbilical sao uma alternativa em recém-nascidos e lactentes.
A farmacoterapia tem algumas particularidades nesta faixa etária. Andrógenos atenuados devem ser evitados, uma vez que provocam o fechamento prematuro da placa epifisária de crescimento, reduzindo a estatura final do paciente. Agentes antifibrinolíticos sao alternativas mais aceitáveis para a profilaxia de longo prazo em crianças com AEH, embora sua eficácia nao tenha sido comprovada e há a possibilidade de ocorrência de eventos adversos. O plasma fresco congelado pode ser utilizado nas crises ou na prevençao de crises associadas a procedimentos cirúrgicos e dentais, quando o concentrado de pdC1-INH nao estiver disponível150. No Brasil, o concentrado de pdC1-INH (20 UI/kg) e o Icatibanto (30 mg) foram aprovados para o tratamento de episódios de AEH em pacientes de 12 e 18 anos de idade, respectivamente.
AEH durante a gravidez, parto, pós-parto e lactaçao
Os hormônios, em particular o estrogênio, sao fatores relevantes de desencadeamento de crises de AEH em pacientes do sexo feminino. Assim, os sintomas podem se tornar mais frequentes e mais graves durante a gravidez, pós-parto e durante a lactaçao em mulheres com AEH com ou sem deficiência de C1-INH64,155,160,161. No entanto, ainda há número limitado de estudos nessas situaçoes, especialmente nos casos de AEH sem deficiência de C1-INH. Devido ao potencial agravamento da doença durante a gravidez, parto, pós-parto e lactaçao, e, também, à restriçao ao uso de medicamentos apropriados, recomendase que as mulheres com AEH sejam acompanhadas de forma frequente e regular durante estas fases de suas vidas64,155.
No planejamento da gravidez, mulheres utilizando profilaxia de longo prazo com andrógenos atenuados devem interromper o tratamento pelo menos um mês antes da concepçao. O uso de andrógenos nao é recomendado durante a gravidez; eles atravessam a barreira placentária e podem resultar em virilizaçao do feto, levando a pseudo-hermafroditismo feminino. O ácido tranexâmico também atravessa a barreira placentária e pode provocar efeitos colaterais para o feto, embora muito menores do que aqueles causados por andrógenos. Devido a sua meia-vida curta, a interrupçao do tratamento com antifibrinolíticos pode ser feita alguns dias antes da concepçao64,155,162.
A gravidez pode melhorar, piorar ou nao ter impacto sobre a frequência e gravidade dos episódios de AEH, o que torna difícil prever a evoluçao das pacientes163-165. Um estudo, que avaliou o prognóstico do AEH com deficiência de C1-INH durante a gravidez, observou um aumento da frequência e gravidade das crises em mulheres grávidas que referiam início precoce dos sintomas de AEH ou que relatavam o trauma como um importante fator desencadeante165,166. A frequência de crises durante gravidez anterior nao tem valor preditivo para a evoluçao do AEH durante gestaçoes posteriores. Por outro lado, pacientes sintomáticas têm uma chance maior de trabalho de parto prematuro ou aborto devido à atividade da bradicinina, que leva à contraçao do músculo liso do útero64. Apesar dos resultados divergentes, a tendência é o agravamento dos sintomas durante o primeiro trimestre da gravidez, quando as mulheres que estavam utilizando profilaxia de longo prazo têm que interromper ou modificar a profilaxia e quando os níveis de estrogênio no sangue sao mais elevados. O segundo trimestre foi descrito como o período mais calmo devido aos níveis permanentemente altos de hormônios. No terceiro trimestre, o aumento dos níveis de progestágenos associado com a produçao de estrógenos placentários e de prolactogênio podem aumentar a atividade da doença64.
Nos casos graves de AEH, quando a profilaxia de longo prazo é necessária, o concentrado de pdC1-INH está indicado6,80,96,167. A dosagem administrada é determinada de acordo com a gravidade da doença e varia de 500 U, uma vez por semana, a 2.000 U, duas vezes por semana168,169. A meia-vida curta (72 horas) e o custo alto do produto ainda dificultam sua utilizaçao rotineira em alguns países. Existem poucos estudos sobre a eficácia do concentrado de pdC1-INH em pacientes com AEH com C1-INH normal e os resultados sao variáveis128,170. O uso de ácido tranexâmico durante a gravidez pode ser indicado quando o concentrado de pdC1-INH nao estiver disponível. A dosagem é semelhante à dose prescrita para pacientes que nao estao grávidas. Alguns autores recomendam o uso com cautela devido ao efeito pró-trombótico, especialmente em pacientes com história pessoal e/ou familiar de tromboembolismo91.
A profilaxia de curto prazo em qualquer procedimento realizado durante a gravidez deverá ser considerada. O tratamento de escolha também é a administraçao de concentrado de pdC1-INH, 1 a 6 horas antes do procedimento64. Se o concentrado nao estiver disponível, plasma fresco congelado e/ou o ácido tranexâmico podem ser administrados. Para evitar uma possível crise durante o parto, alguns autores têm recomendado a administraçao de concentrado de pdC1-INH (500 ou 1.000 U), uma hora antes do procedimento163,165. No entanto, um consenso internacional recomendou abordagem de observaçao durante o parto, assim como a disponibilidade de, pelo menos, uma dose de concentrado de pdC1-INH na sala de parto. A administraçao profilática de concentrado de pdC1-INH é formalmente indicada para partos difíceis exigindo fórceps, em pacientes sem controle da doença e em caso de cesariana64.
Durante a gravidez, as crises se manifestam predominantemente no abdômen, o que dificulta o diagnóstico diferencial com outras complicaçoes associadas à gravidez64. A ultrassonografia abdominal é útil no diagnóstico diferencial nessa situaçao. O tratamento de crises durante a gravidez inclui a prescriçao de medicamentos sintomáticos (analgésicos), hidrataçao e uso de concentrado de pdC1-INH ou plasma fresco congelado, quando o primeiro nao estiver disponível64.
Crises durante o parto geralmente ocorrem imediatamente após ou no prazo de 48 horas posteriores ao mesmo, e podem ter consequências graves64,171. No período pós-parto, algumas mulheres podem experimentar angioedema da vulva e dos locais de infusao, bem como obstruçao uretral e crises abdominais, sendo, portanto, recomendada a observaçao da paciente durante 72 horas após o parto6,64. Estudos mostram que, independentemente do tipo de parto, as crises sao raras, mesmo sem profilaxia164,165. O hospital onde o parto será realizado deve ter medicaçao de prevençao e pessoal capacitado para assistência de pacientes com AEH.
A maioria das mulheres experimenta aumento da frequência e da gravidade das crises de AEH durante a lactaçao, interferindo com a amamentaçao172. Concentraçoes mais elevadas de prolactina parecem ser responsáveis pelo aumento temporário das crises após o parto165. Andrógenos e antifibrinolíticos sao excretados no leite humano e, por isso, devem ser evitados durante a amamentaçao.
O tratamento de escolha das crises durante a gravidez, parto, pós-parto e amamentaçao é o concentrado de pdC1-INH. Vários estudos têm comprovado a sua eficácia e segurança64,163,165. Quando o concentrado de pdC1-INH nao estiver disponível para tratamento das crises, o plasma fresco congelado pode ser administrado64. Para ambos, a dosagem, os riscos e as precauçoes sao os mesmos que para as mulheres nao grávidas. Nao há estudos controlados de tratamento de episódios de AEH durante esses períodos com Icatibanto, Ecallantide e C1-INH recombinante.
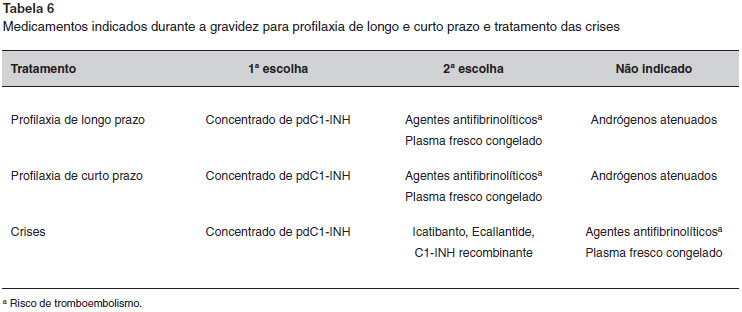
Em resumo, a utilizaçao dos medicamentos disponíveis para o tratamento do AEH com ou sem deficiência de C1-INH é limitada durante a gravidez, o parto ou pós-parto (Tabela 6). O concentrado de pdC1-INH é o único medicamento recomendado por todos os estudos publicados. Em nosso país, o concentrado de pdC1-INH atualmente é aprovado pela ANVISA, embora ainda nao esteja incluído na lista de medicamentos de alto custo fornecidos pelo governo.
QUAL É O IMPACTO DO ANGIOEDEMA HEREDITARIO SOBRE A QUALIDADE DE VIDA?
Ao longo das últimas três décadas, estudos têm cada vez mais avaliado o impacto de várias doenças sobre a qualidade de vida dos pacientes (QV), mas só recentemente foi estudado o impacto do AEH sobre a QV dos pacientes8. As publicaçoes ainda sao escassas e, somente há pouco, foi padronizado um questionário internacional específico a partir de um projeto multicêntrico espanhol173.
Um estudo brasileiro com 35 pacientes com idade superior a 15 anos, utilizando o Short Form-36 (SF-36), observou que 90,4% tiveram pontuaçao abaixo de 70, sendo a vitalidade e os aspectos sociais as áreas mais afetadas84. Em uma coorte sueca, foi observada reduçao significativa na pontuaçao EuroQol 5D (EQ5D) durante a crise de AEH. Absenteísmo do trabalho/escola foi detectado em 44,6% dos pacientes, sendo mais frequente em pacientes com doença grave (81%)174.
O AEH reduz significativamente a qualidade de vida dos pacientes, a qual pode ser recuperada com o tratamento adequado108,112. Um estudo, utilizando os questionários Dermatology Life Quality Index (DLQI, Indice de Qualidade de Vida em Dermatologia) e SF-36, mostrou que a administraçao intravenosa caseira do concentrado de pdC1-INH melhorou a qualidade de vida de pacientes com doença grave ou debilitante175. O desenvolvimento de novas ferramentas de avaliaçao da QV ajudará os profissionais de saúde a quantificar o nível de reduçao causado pelo AEH e o impacto de intervençoes terapêuticas.
CONSIDERAÇOES FINAIS
A educaçao dos profissionais da área da saúde, dos pacientes e de seus familiares é fundamental para o sucesso da abordagem do AEH, a qual tem melhorado nos últimos anos no Brasil. O reconhecimento de pacientes com a doença tem sido maior e mais precoce, e novas opçoes terapêuticas estao disponíveis agora1. Por outro lado, o acesso à medicaçao ainda é restrito176.
Em relaçao ao diagnóstico do AEH, Bork et al. identificaram subtipos de AEH com C1-INH normal, um deles associado às mutaçoes do fator XII da coagulaçao46,52. Há outras condiçoes que causam o angioedema sem urticária que tem causas genéticas e ambientais desconhecidas, e sao difíceis de serem diagnosticadas e tratadas22.
A profilaxia de longo prazo com pdC1-INH foi estabelecida para os doentes com doença grave ou com condiçoes especiais177. Estudos estao avaliando o uso subcutâneo destes produtos, o que promete ser um avanço no tratamento dos pacientes113. Outros produtos estao sendo investigados para a profilaxia de longo prazo, incluindo um bloqueador de bradicinina e um inibidor da calicreína, ambos de uso oral, e um anticorpo monoclonal que inibe a calicreína do plasma de uso subcutâneo (DX-2930)148,178-181. Como as terapias preventivas nem sempre sao eficazes e nao têm indicaçao universal, os pacientes devem dispor de medicamentos para tratamento das crises de AEH. A autoadministraçao domiciliar de Icatibanto e de pdC1-INH foi aprovada pela Agência Reguladora Brasileira, a ANVISA, estando de acordo com a experiência internacional.
Especialistas da Associaçao Brasileira de Alergia e Imunologia (ASBAI) e do Grupo Brasileiro de Estudos de Angioedema Hereditário (GEBRAEH) desenvolveram essas diretrizes para o diagnóstico e terapia do AEH, com o objetivo de ajudar os profissionais da área da saúde na identificaçao e abordagem desta doença. O AEH atualmente é menos negligenciado, mas precisamos continuar progredindo. Uma versao em inglês destas diretrizes será publicada simultaneamente na revista Clinics.
REFERENCIAS
1. Giavina-Bianchi P, França AT, Grumach AS, Motta AA, Fernandes FR, Campos RA, et al. Brazilian guidelines for the diagnosis and treatment of hereditary angioedema. Clinics (Sao Paulo). 2011;66:1627-36.
2. Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore). 1992;71:206-15.
3. Cicardi M, Bergamaschini L, Marasini B, Boccassini G, Tucci A, Agostoni A. Hereditary angioedema: an appraisal of 104 cases. Am J Med Sci. 1982;284:2-9.
4. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69:602-16.
5. Bork K, Meng G, Staubach P, Hardt J. Hereditary Angioedema: New Findings Concerning Symptoms, Affected Organs, and Course. Am J Med. 2006;119:267-74.
6. Craig T. WAO Guideline for the Management of Hereditary Angioedema. WAO Journal. 2012;5:182-99.
7. Gomide MACMS, Toledo E, Valle SOR, Campos RA, França AT, Gomez NP, et al. Hereditary angioedema: quality of life in Brazilian patients. Clinics (Sao Paulo). 2013;68:81-3.
8. Prior N, Remor E, Perez-Fernandez E, Caminoa M, Gomez-Traseira C, Gaya F, et al. Psychometric Field Study of Hereditary Angioedema Quality of Life Questionnaire for Adults: HAE-QoL. J Allergy Clin Immunol Pract. 2016;4:464-73.e4.
9. Lumry WR, Castaldo AJ, Vernon MK, Blaustein MB, Wilson DA, Horn PT. The humanistic burden of hereditary angioedema: Impact on health-related quality of life, productivity, and depression. Allergy Asthma Proc. 2010;31:407-14.
10. Farkas H, Varga L, Szeplaki G, Visy B, Harmat G, Bowen T. Management of hereditary angioedema in pediatric patients. Pediatrics. 2007;120:e713-22.
11. Lunn ML, Santos CB, Craig TJ. Is there a need for clinical guidelines in the United States for the diagnosis of hereditary angioedema and the screening of family members of affected patients? Ann Allergy Asthma Immunol. 2010;104:211-4.
12. Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114(3 Suppl):S51-131.
13. Longhurst HJ, Farkas H, Craig T, Aygören-Pürsün E, Bethune C, Bjorkander J, et al. HAE international home therapy consensus document. Allergy Asthma Clin Immunol. 2010;6:22.
14. Bowen T, Cicardi M, Farkas H, Bork K, Longhurst HJ, Zuraw B, et al. 2010 International consensus algorithm for the diagnosis, therapy and management of hereditary angioedema. Allergy Asthma Clin Immunol. 2010;6:24.
15. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356:213-7.
16. Cicardi M, Bergamaschini L, Marasini B, Boccassini G, Tucci A, Agostoni A. Hereditary angioedema: an appraisal of 104 cases. Am J Med Sci. 1982;284:2-9.
17. Bork K, Hardt J, Schicketanz K-H, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med. 2003;163:1229-35.
18. Bork K. Diagnosis and treatment of hereditary angioedema with normal C1 inhibitor. Allergy Asthma Clin Immunol. 2010;6:15.
19. Moore GP, Hurley WT, Pace SA. Hereditary angioedema. Ann Emerg Med. 1988;17:1082-6.
20. Farkas H, Harmat G, Kaposi PN, Karadi I, Fekete B, Fust G, et al. Ultrasonography in the diagnosis and monitoring of ascites in acute abdominal attacks of hereditary angioneurotic oedema. Eur J Gastroenterol Hepatol. 2001;13:1225-30.
21. Bork K, Staubach P, Eckardt AJ, Hardt J. Symptoms, course, and complications of abdominal attacks in hereditary angioedema due to C1 inhibitor deficiency. Am J Gastroenterol. 2006;101:619-27.
22. Giavina-Bianchi P, Aun MV, Motta AA, Kalil J, Castells M. Classification of angioedema by endotypes. Clin Exp Allergy. 2015;45:1142-3.
23. Quincke HI. Über akutes umschriebenes Hautödem. Monatsh Prakt Dermatol. 1982;1:3.
24. Giavina-Bianchi P, Aun MV, Jares EJ, Kalil J. Angioedema associated with nonsteroidal anti-inflammatory drugs. Curr Opin Allergy Clin Immunol. 2016;16:323-32.
25. Aun MV, Blanca M, Garro LS, Ribeiro MR, Kalil J, Motta AA, et al. Nonsteroidal Anti-Inflammatory Drugs are Major Causes of Drug-Induced Anaphylaxis. J Allergy Clin Immunol Pract. 2014;2:414-20.
26. Osler W. Hereditary angioneurotic edema. Am J Med Sci. 1888;95:6.
27. Gomes E, Cardoso MF, Praca F, Gomes L, Marino E, Demoly P. Self-reported drug allergy in a general adult Portuguese population. Clin Exp Allergy. 2004;34:1597-601.
28. Stevenson DD, Sanchez-Borges M, Szczeklik A. Classification of allergic and pseudoallergic reactions to drugs that inhibit cyclooxygenase enzymes. Ann Allergy Asthma Immunol. 2001;87:177-80.
29. Kowalski ML, Asero R, Bavbek S, Blanca M, Blanca-Lopez N, Bochenek G, et al. Classification and practical approach to the diagnosis and management of hypersensitivity to nonsteroidal anti-inflammatory drugs. Allergy. 2013;68:1219-32.
30. Landerman NS, Webster ME, Becker EL, Ratcliffe HE. Hereditary angioneurotic edema. II. Deficiency of inhibitor for serum globulin permeability factor and/or plasma kallikrein. J Allergy. 1962;33:330-41.
31. Donaldson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C'1-esterase. Am J Med. 1963;35:37-44.
32. Rosen FS, Pensky J, Donaldson V, Charache P. Hereditary angioneurotic edema: two genetic variants. Science. 1965;148:957-8.
33. Dunkelberger JR, Song W-C. Complement and its role in innate and adaptive immune responses. Cell Research. 2010;20:34-50.
34. Ratnoff OD, Pensky J, Ogston D, Naff GB. The inhibition of plasmin, plasma kallikrein, plasma permeability factor, and the C"1r subcomponent of the first component of complement by serum C"1 esterase inhibitor. J Exp Med. 1969;129:315-31.
35. Bork K, Witzke G, Artmann K, Benes P, Bockers M, Kreuz W. Interaction between C1-INA, coagulation, fibrinolysis and kinin system in hereditary angioneurotic edema (HANE) and urticaria. Arch Dermatol Res. 1984;276:375-80.
36. Walford HH, Zuraw BL. Current update on cellular and molecular mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112:413-8.
37. Rocha e Silva M, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am J Physiol. 1949;156:261-73.
38. Elliott DF, Horton EW, Lewis GP. Actions of pure bradykinin. J Physiol. 1960;153:473-80.
39. Venema VJ, Marrero MB, Venema RC. Bradykinin-stimulated protein tyrosine phosphorylation promotes endothelial nitric oxide synthase translocation to the cytoskeleton. Biochem Biophys Res Commun. 1996;226:703-10.
40. Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351:1693-7.
41. Nussberger J, Cugno M, Cicardi M. Bradykinin-mediated angioedema. N Engl J Med. 2002;347:621-2.
42. Nussberger J, Cugno M, Cicardi M, Agostoni A. Local bradykinin generation in hereditary angioedema. J Allergy Clin Immunol. 1999;104:1321-2.
43. Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE III. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109:1057-63.
44. Cicardi M, Banerji A, Bracho F, Malbran A, Rosenkranz B, Riedl M, et al. Icatibant, a New Bradykinin-Receptor Antagonist, in Hereditary Angioedema. N Engl J Med. 2010;363:532-41.
45. Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, Horn PT, Pullman WE. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-31.
46. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356:213-7.
47. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343:1286-9.
48. Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14:28-39.
49. Cichon S, Martin L, Hennies HC, Muller F, Van Driessche K, Karpushova A, et al. Increased activity of coagulation factor XII (Hageman factor) causes hereditary angioedema type III. Am J Hum Genet. 2006;79:1098-104.
50. Bjorkqvist J, de Maat S, Lewandrowski U, Di Gennaro A, Oschatz C, Schonig K, et al. Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J Clin Invest. 2015;125:3132-46.
51. de Maat S, Bjorkqvist J, Suffritti C, Wiesenekker CP, Nagtegaal W, Koekman A, et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. 2016;138:1414-9.
52. Bork K, Wulff K, Witzke G, Hardt J. Hereditary angioedema with normal C1-INH with versus without specific F12 gene mutations. Allergy. 2015;70:1004-12.
53. Bork K. Hereditary angioedema with normal C1 inhibitor. Immunol Allergy Clin North Am. 2013;33:457-70.
54. Citarella F, Misiti S, Felici A, Aiuti A, La Porta C, Fantoni A. The 5' sequence of human factor XII gene contains transcription regulatory elements typical of liver specific, estrogen-modulated genes. Biochim Biophys Acta. 1993;1172:197-9.
55. Madeddu P, Emanueli C, Song Q, Varoni MV, Demontis MP, Anania V, et al. Regulation of bradykinin B2-receptor expression by oestrogen. Br J Pharmacol. 1997;121:1763-9.
56. Chen LM, Chung P, Chao S, Chao L, Chao J. Differential regulation of kininogen gene expression by estrogen and progesterone in vivo. Biochim Biophys Acta. 1992;1131:145-51.
57. Zuraw BL, Herschbach J. Detection of C1 inhibitor mutations in patients with hereditary angioedema. J Allergy Clin Immunol. 2000;105:541-6.
58. Bowen B, Hawk JJ, Sibunka S, Hovick S, Weiler JM. A review of the reported defects in the human C1 esterase inhibitor gene producing hereditary angioedema including four new mutations. Clin Immunol. 2001;98:157-63.
59. Matesic D, Fernandez Perez ER, Vlahakis NE, Hagan JB. Acute pancreatitis due to hereditary angioedema. Ann Allergy Asthma Immunol. 2006;97:611-4.
60. Li HH, Busse P, Lumry WR, Frazer-Abel A, Levy H, Steele T, et al. Comparison of chromogenic and ELISA functional C1 inhibitor tests in diagnosing hereditary angioedema. J Allergy Clin Immunol Pract. 2015;3:200-5.
61. Wagenaar-Bos IGA, Drouet C, Aygören-Pürsün E, Bork K, Bucher C, Bygum A, et al. Functional C1-inhibitor diagnostics in hereditary angioedema: assay evaluation and recommendations. J Immunol Methods. 2008;338:14-20.
62. Farkas H, Jakab L, Temesszentandrasi G, Visy B, Harmat G, Fust G, et al. Hereditary angioedema: a decade of human C1-inhibitor concentrate therapy. J Allergy Clin Immunol. 2007;120:941-7.
63. Weiler CR, van Dellen RG. Genetic test indications and interpretations in patients with hereditary angioedema. Mayo Clin Proc. 2006;81:958-72.
64. Caballero T, Farkas H, Bouillet L, Bowen T, Gompel A, Fagerberg C, et al. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012;129:308-20.
65. Kiss N, Barabas E, Varnai K, Halasz A, Varga LA, Prohaszka Z, et al. Novel duplication in the F12 gene in a patient with recurrent angioedema. Clin Immunol. 2013;149:142-5.
66. Bork K, Wulff K, Meinke P, Wagner N, Hardt J, Witzke G. A novel mutation in the coagulation factor 12 gene in subjects with hereditary angioedema and normal C1-INHibitor. Clin Immunol. 2011;141:31-5.
67. Farkas H, Veszeli N, Kajdacsi E, Cervenak L, Varga L. "Nuts and Bolts" of Laboratory Evaluation of Angioedema. Clin Rev Allergy Immunol. 2016;51:140-51.
68. Zuberbier T, Aberer W, Asero R, Bindslev-Jensen C, Brzoza Z, Canonica GW, et al. The EAACI/GA(2) LEN/EDF/WAO Guideline for the definition, classification, diagnosis, and management of urticaria: the 2013 revision and update. Allergy. 2014;69:868-87.
69. Miller DR, Oliveria SA, Berlowitz DR, Fincke BG, Stang P, Lillienfeld DE. Angioedema incidence in US veterans initiating angiotensin-converting enzyme inhibitors. Hypertension. 2008;51:1624-30.
70. Kostis JB, Kim HJ, Rusnak J, Casale T, Kaplan A, Corren J, et al. Incidence and characteristics of angioedema associated with enalapril. Arch Intern Med. 2005;165:1637-42.
71. Beltrami L, Zanichelli A, Zingale L, Vacchini R, Carugo S, Cicardi M. Long-term follow-up of 111 patients with angiotensin-converting enzyme inhibitor-related angioedema. J Hypertens. 2011;29:2273-7.
72. Makani H, Messerli FH, Romero J, Wever-Pinzon O, Korniyenko A, Berrios RS, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol. 2012;110:383-91.
73. Schreiber AD, Zweiman B, Atkins P, Goldwein F, Pietra G, Atkinson B, et al. Acquired angioedema with lymphoproliferative disorder: association of C1 inhibitor deficiency with cellular abnormality. Blood. 1976;48:567-80.
74. Mandle R, Baron C, Roux E, Sundel R, Gelfand J, Aulak K, et al. Acquired C1 inhibitor deficiency as a result of an autoantibody to the reactive center region of C1 inhibitor. J Immunol. 1994;152:4680-5.
75. Zingale LC, Castelli R, Zanichelli A, Cicardi M. Acquired deficiency of the inhibitor of the first complement component: presentation, diagnosis, course, and conventional management. Immunol Allergy Clin North Am. 2006;26:669-90.
76. Zuraw BL, Bernstein JA, Lang DM, Craig T, Dreyfus D, Hsieh F, et al. A focused parameter update: hereditary angioedema, acquired C1 inhibitor deficiency, and angiotensin-converting enzyme inhibitor-associated angioedema. J Allergy Clin Immunol. 2013;131:1491-3.
77. Zuraw BL, Curd JG. Demonstration of modified inactive first component of complement (C1) inhibitor in the plasmas of C1 inhibitor-deficient patients. J Clin Invest. 1986;78:567-75.
78. Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore). 1992;71:206-15.
79. Bork K, Hardt J, Schicketanz K-H, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency. Arch Intern Med. 2003;163:1229-35.
80. Costantino G, Casazza G, Bossi I, Duca P, Cicardi M. Long-term prophylaxis in hereditary angio-oedema: a systematic review. BMJ Open. 2012;2(4).
81. Bork K, Witzke G. Long-term prophylaxis with C1-inhibitor (C1 INH) concentrate in patients with recurrent angioedema caused by hereditary and acquired C1-inhibitor deficiency. J Allergy Clin Immunol. 1989;83:677-82.
82. Bork K, Hardt J. Hereditary angioedema: long-term treatment with one or more injections of C1 inhibitor concentrate per week. Int Arch Allergy Immunol. 2011;154:81-8.
83. Zuraw BL. Hereditary angiodema: a current state-of-the-art review, IV: short- and long-term treatment of hereditary angioedema: out with the old and in with the new? Ann Allergy Asthma Immunol. 2008;100(1 Suppl 2):S13-8.
84. Gomide MACMS, Toledo E, Valle SOR, Campos RA, França AT, Gomez NP, et al. Hereditary angioedema: quality of life in Brazilian patients. Clinics. 2013;68:81-3.
85. CaballeroT, Aygören-Pürsün E, Bygum A, Beusterien K, Hautamaki E, Sisic Z, et al. The humanistic burden of hereditary angioedema: results from the Burden of Illness Study in Europe. Allergy Asthma Proc. 2014;35:47-53.
86. Zuraw BL, Cicardi M, Longhurst HJ, Bernstein JA, Li HH, Magerl M, et al. Phase II study results of a replacement therapy for hereditary angioedema with subcutaneous C1-INHibitor concentrate. Allergy. 2015;70:1319-28.
87. Nilsson IM, Andersson L, Bjorkman SE. Epsilon-aminocaproic acid (E-ACA) as a therapeutic agent based on 5 year's clinical experience. Acta Med Scand Suppl. 1966;448:1-46.
88. Frank MM, Sergent JS, Kane MA, Alling DW. Epsilon aminocaproic acid therapy of hereditary angioneurotic edema. A double-blind study. N Engl J Med. 1972;286:808-12.
89. Wintenberger C, Boccon-Gibod I, Launay D, Fain O, Kanny G, Jeandel PY, et al. Tranexamic acid as maintenance treatment for non-histaminergic angioedema: analysis of efficacy and safety in 37 patients. Clin Exp Immunol. 2014;178:112-7.
90. Tengborn L, Blomback M, Berntorp E. Tranexamic acid - an old drug still going strong and making a revival. Thromb Res. 2015;135:231-42.
91. Gompels MM, Lock RJ, Abinun M, Bethune CA, Davies G, Grattan C, et al. C1 inhibitor deficiency: consensus document. Clin Exp Immunol. 2005;139:379-94.
92. Levy JH, Freiberger DJ, Roback J. Hereditary angioedema: current and emerging treatment options. Anesth Analg. 2010;110:1271-80.
93. Blohme G. Treatment of hereditary angioneurotic oedema with tranexamic acid. A random double-blind cross-over study. Acta Med Scand. 1972;192:293-8.
94. Sheffer AL, Austen KF, Rosen FS. Tranexamic acid therapy in hereditary angioneurotic edema. N Engl J Med. 1972;287:452-4.
95. Frank MM. Update on preventive therapy (prophylaxis) for hereditary angioedema. Immunol Allergy Clin North Am. 2013;33:495-503.
96. Cicardi M, Bork K, Caballero T, Craig T, Li HH, Longhurst H, et al. Evidence-based recommendations for the therapeutic management of angioedema owing to hereditary C1 inhibitor deficiency: consensus report of an International Working Group. Allergy. 2012;67:147-57.
97. D'Ottaviano FL, Tanno LK, Kalil J, Motta AA, Giavina-Bianchi P. Clinical Features and Outcome of 52 Patients with Hereditary Angioedema. J Allergy Clin Immunol. 2011;127:AB233-3.
98. Cicardi M, Castelli R, Zingale LC, Agostoni A. Side effects of long-term prophylaxis with attenuated androgens in hereditary angioedema: comparison of treated and untreated patients. J Allergy Clin Immunol. 1997;99:194-6.
99. Szeplaki G, Varga L, Valentin S, Kleiber M, Karadi I, Romics L, et al. Adverse effects of danazol prophylaxis on the lipid profiles of patients with hereditary angioedema. J Allergy Clin Immunol. 2005;115:864-9.
100. Birjmohun RS, Kees Hovingh G, Stroes ESG, Hofstra JJ, Dallinga-Thie GM, Meijers JCM, et al. Effects of short-term and long-term danazol treatment on lipoproteins, coagulation, and progression of atherosclerosis: two clinical trials in healthy volunteers and patients with hereditary angioedema. CLITHE. 2008;30:2314-23.
101. Zurlo JJ, Frank MM. The long-term safety of danazol in women with hereditary angioedema. Fertil Steril. 1990;54:64-72.
102. Bork K, Pitton M, Harten P, Koch P. Hepatocellular adenomas in patients taking danazol for hereditary angio-oedema. Lancet. 1999;353:1066-7.
103. Monnier N, Ponard D, Duponchel C, Csopaki F, Bouillet L, Tosi M, et al. Characterisation of a new C1 inhibitor mutant in a patient with hepatocellular carcinoma. Mol Immunol. 2006;43:2161-8.
104. Berkel AEM, Bouman DE, Schaafsma MR, Verhoef C, Klaase JM. Hepatocellular carcinoma after danazol treatment for hereditary angio-oedema. Neth J Med. 2014;72:380-2.
105. Farkas H, Czaller I, Csuka D, Vas A, Valentin S, Varga L, et al. The effect of long-term danazol prophylaxis on liver function in hereditary angioedema-a longitudinal study. Eur J Clin Pharmacol. 2010;66:419-26.
106. Waytes AT, Rosen FS, Frank MM. Treatment of hereditary angioedema with a vapor-heated C1 inhibitor concentrate. N Engl J Med. 1996;334:1630-4.
107. Tallroth GA. Long-term prophylaxis of hereditary angioedema with a pasteurized C1 inhibitor concentrate. Int Arch Allergy Immunol. 2011;154:356-9.
108. Kreuz W, Martinez-Saguer I, Aygören-Pürsün E, Rusicke E, Heller C, Klingebiel T. C1-inhibitor concentrate for individual replacement therapy in patients with severe hereditary angioedema refractory to danazol prophylaxis. Transfusion. 2009;49:1987-95.
109. Zuraw BL, Busse PJ, White M, Jacobs J, Lumry W, Baker J, et al. Nanofiltered C1 inhibitor concentrate for treatment of hereditary angioedema. N Engl J Med. 2010;363:513-22.
110. Frank MM. Hereditary angiodema: a current state-of-the-art review, VI: novel therapies for hereditary angioedema. Ann Allergy Asthma Immunol. 2008;100(1 Suppl 2):S23-9.
111. Gandhi PK, Gentry WM, Bottorff MB. Thrombotic events associated with C1 esterase inhibitor products in patients with hereditary angioedema: investigation from the United States Food and Drug Administration adverse event reporting system database. Pharmacotherapy. 2012;32:902-9.
112. Dagen C, Craig TJ. Treatment of Hereditary Angioedema: items that need to be addressed in practice parameter. Allergy Asthma Clin Immunol. 2010;6:11.
113. Zuraw BL, Cicardi M, Longhurst HJ, Bernstein JA, Li HH, Magerl M, et al. Phase II study results of a replacement therapy for hereditary angioedema with subcutaneous C1-INHibitor concentrate. Allergy. 2015;70:1319-28.
114. Reshef A, Moldovan D, Obtulowicz K, Leibovich I, Mihaly E, Visscher S, et al. Recombinant human C1 inhibitor for the prophylaxis of hereditary angioedema attacks: a pilot study. Allergy. 2013;68:118-24.
115. Deroux A, Boccon-Gibod I, Fain O, Pralong P, Ollivier Y, Pagnier A, et al. Hereditary angioedema with normal C1 inhibitor and factor XII mutation: a series of 57 patients from the French National Center of Reference for Angioedema. Clin Exp Immunol. 2016;185:332-7.
116. Bernstein JA. Managing hereditary angioedema patients undergoing otolaryngeal procedures. Am J Rhinol Allergy. 2013;27:522-7.
117. Aygoren-Pursun E, Martinez-Saguer I, Kreuz W, Klingebiel T, Schwabe D. Risk of angioedema following invasive or surgical procedures in HAE type I and II - the natural history. Allergy. 2013;68:1034-9.
118. Jurado-Palomo J, Munoz-Caro JM, Lopez-Serrano MC, Prior N, Cabanas R, Pedrosa M, et al. Management of dental-oral procedures in patients with hereditary angioedema due to C1 inhibitor deficiency. J Investig Allergol Clin Immunol. 2013;23:1-6.
119. Bork K, Hardt J, Staubach-Renz P, Witzke G. Risk of laryngeal edema and facial swellings after tooth extraction in patients with hereditary angioedema with and without prophylaxis with C1 inhibitor concentrate: a retrospective study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112:58-64.
120. Bork K. An evidence based therapeutic approach to hereditary and acquired angioedema. Current Opinion in Allergy and Clinical Immunology. 2014;14:354-62.
121. Farkas H, Zotter Z, Csuka D, Szabo E, Nebenfuhrer Z, Temesszentandrasi G, et al. Short-term prophylaxis in hereditary angioedema due to deficiency of the C1-inhibitor - a long-term survey. Allergy. 2012;67:1586-93.
122. Galan HL, Reedy MB, Starr J, Knight AB. Fresh frozen plasma prophylaxis for hereditary angioedema during pregnancy. A case report. J Reprod Med. 1996;41:541-4.
123. Birjmohun RS, Kees Hovingh G, Stroes ESG, Hofstra JJ, Dallinga-Thie GM, Meijers JCM, et al. Effects of short-term and long-term danazol treatment on lipoproteins, coagulation, and progression of atherosclerosis: two clinical trials in healthy volunteers and patients with hereditary angioedema. CLITHE. 2008;30:2314-23.
124. Banerji A, Busse P, Christiansen SC, Li H, Lumry W, Davis-Lorton M, et al. Current state of hereditary angioedema management: a patient survey. Allergy Asthma Proc. 2015;36:213-7.
125. Christiansen SC, Bygum A, Banerji A, Busse P, Li H, Lumry W, et al. Before and after, the impact of available on-demand treatment for HAE. Allergy Asthma Proc. 2015;36:145-50.
126. Prematta M, Gibbs JG, Pratt EL, Stoughton TR, Craig TJ. Fresh frozen plasma for the treatment of hereditary angioedema. Ann Allergy Asthma Immunol. 2007;98:383-8.
127. Nzeako UC, Frigas E, Tremaine WJ. Hereditary angioedema: a broad review for clinicians. Arch Intern Med. 2001;161:2417-29.
128. Zuraw BL, Banerji A, Bernstein JA, Busse PJ, Christiansen SC, Davis-Lorton M, et al. US Hereditary Angioedema Association Medical Advisory Board 2013 recommendations for the management of hereditary angioedema due to C1 inhibitor deficiency. J Allergy Clin Immunol Pract. 2013;1:458-67.
129. Bork K, Bygum A, Hardt J. Benefits and risks of danazol in hereditary angioedema: a long-term survey of 118 patients. Ann Allergy Asthma Immunol. 2008;100:153-61.
130. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130:692-7.
131. Craig TJ, Bewtra AK, Bahna SL, Hurewitz D, Schneider LC, Levy RJ, et al. C1 esterase inhibitor concentrate in 1085 Hereditary Angioedema attacks - final results of the I.M.P.A.C.T.2 study. Allergy. 2011;66:1604-11.
132. Craig TJ, Levy RJ, Wasserman RL, Bewtra AK, Hurewitz D, Obtulowicz K, et al. Efficacy of human C1 esterase inhibitor concentrate compared with placebo in acute hereditary angioedema attacks. J Allergy Clin Immunol. 2009;124:801-8.
133. Bork K. Pasteurized and nanofiltered, plasma-derived C1 esterase inhibitor concentrate for the treatment of hereditary angioedema. Immunotherapy. 2014;6:533-51.
134. Bork K, Meng G, Staubach P, Hardt J. Treatment with C1 inhibitor concentrate in abdominal pain attacks of patients with hereditary angioedema. Transfusion. 2005;45:1774-84.
135. Bork K, Staubach P, Hardt J. Treatment of skin swellings with C1-inhibitor concentrate in patients with hereditary angio-oedema. Allergy. 2008;63:751-7.
136. Bork K, Barnstedt SE. Treatment of 193 episodes of laryngeal edema with C1 inhibitor concentrate in patients with hereditary angioedema. Arch Intern Med. 2001;161:714-8.
137. Bhardwaj N, Craig TJ. Treatment of hereditary angioedema: a review (CME). Transfusion. 2014;54:2989-96-quiz2988.
138. Farkas H, Varga L. Human Plasma-Derived, Nanofiltered, C1-Inhibitor Concentrate (Cinryze(R)), a Novel Therapeutic Alternative for the Management of Hereditary Angioedema Resulting from C1-Inhibitor Deficiency. Biol Ther. 2012;2:2.
139. Zuraw B, Cicardi M, Levy RJ, Nuijens JH, Relan A, Visscher S, et al. Recombinant human C1-inhibitor for the treatment of acute angioedema attacks in patients with hereditary angioedema. J Allergy Clin Immunol. 2010;126:821-827.e14.
140. Riedl MA, Bernstein JA, Li H, Reshef A, Lumry W, Moldovan D, et al. Recombinant human C1-esterase inhibitor relieves symptoms of hereditary angioedema attacks: phase 3, randomized, placebo-controlled trial. Ann Allergy Asthma Immunol. 2014;112:163-9 e1.
141. Hemperly SE, Agarwal NS, Xu Y-Y, Zhi Y-X, Craig TJ. Recent advances in the management of hereditary angioedema. J Am Osteopath Assoc. 2013;113:546-55.
142. Cicardi M, Banerji A, Bracho F, Malbran A, Rosenkranz B, Riedl M, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363:532-41.
143. Lumry WR, Li HH, Levy RJ, Potter PC, Farkas H, Moldovan D, et al. Randomized placebo-controlled trial of the bradykinin B(2) receptor antagonist icatibant for the treatment of acute attacks of hereditary angioedema: the FAST-3 trial. Ann Allergy Asthma Immunol. 2011;107:529-37.
144. Maurer M, Aberer W, Bouillet L, Caballero T, Fabien V, Kanny G, et al. Hereditary angioedema attacks resolve faster and are shorter after early icatibant treatment. PLoS ONE. 2013;8:e53773.
145. Aberer W, Maurer M, Reshef A, Longhurst H, Kivity S, Bygum A, et al. Open-label, multicenter study of self-administered icatibant for attacks of hereditary angioedema. Allergy. 2014;69:305-14.
146. Lumry WR, Farkas H, Moldovan D, Toubi E, Baptista J, Craig T, et al. Icatibant for Multiple Hereditary Angioedema Attacks across the Controlled and Open-Label Extension Phases of FAST-3. Int Arch Allergy Immunol. 2015;168:44-55.
147. Bernstein JA, Qazi M. Ecallantide: its pharmacology, pharmacokinetics, clinical efficacy and tolerability. Expert Rev Clin Immunol. 2010;6:29-39.
148. Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363:523-31.
149. Sheffer AL, Campion M, Levy RJ, Li HH, Horn PT, Pullman WE. Ecallantide (DX-88) for acute hereditary angioedema attacks: integrated analysis of 2 double-blind, phase 3 studies. J Allergy Clin Immunol. 2011;128:153-4.
150. MacGinnitie AJ. Pediatric hereditary angioedema. Pediatr Allergy Immunol. 2014;25:420-7.
151. Donaldson VH, Rosen FS. Hereditary angioneurotic edema: a clinical survey. Pediatrics. 1966;37:1017-27.
152. Farkas H. Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma Clin Immunol. 2010;6:18.
153. Bygum A, Fagerberg CR, Ponard D, Monnier N, Lunardi J, Drouet C. Mutational spectrum and phenotypes in Danish families with hereditary angioedema because of C1 inhibitor deficiency. Allergy. 2011;66:76-84.
154. Roche O, Blanch A, Caballero T, Sastre N, Callejo D, Lopez-Trascasa M. Hereditary angioedema due to C1 inhibitor deficiency: patient registry and approach to the prevalence in Spain. Ann Allergy Asthma Immunol. 2005;94:498-503.
155. Farkas H, Martinez-Saguer I, Bork K, Bowen T, Craig T, Frank M, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy. 2017;72:300-13.
156. Read N, Lim E, Tarzi MD, Hildick-Smith P, Burns S, Fidler KJ. Paediatric hereditary angioedema: a survey of UK service provision and patient experience. Clin Exp Immunol. 2014;178:483-8.
157. Pedrosa M, Phillips-Angles E, Lopez-Lera A, Lopez-Trascasa M, Caballero T. Complement Study Versus CINH Gene Testing for the Diagnosis of Type I Hereditary Angioedema in Children. J Clin Immunol. 2016;36:16-8.
158. Grumach AS, Ceccon ME, Rutz R, Fertig A, Kirschfink M. Complement profile in neonates of different gestational ages. Scand J Immunol. 2014;79:276-81.
159. Nielsen EW, Johansen HT, Holt J, Mollnes TE. C1 inhibitor and diagnosis of hereditary angioedema in newborns. Pediatr Res. 1994;35:184-7.
160. Bork K, Wulff K, Hardt J, Witzke G, Staubach P. Hereditary angioedema caused by missense mutations in the factor XII gene: clinical features, trigger factors, and therapy. J Allergy Clin Immunol. 2009;124:129-34.
161. Zuraw BL, Bork K, Binkley KE, Banerji A, Christiansen SC, Castaldo A, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33 Suppl 1:S145-56.
162. Cicardi M, Bergamaschini L, Cugno M, Hack E, Agostoni G, Agostoni A. Long-term treatment of hereditary angioedema with attenuated androgens: a survey of a 13-year experience. J Allergy Clin Immunol. 1991;87:768-73.
163. Martinez-Saguer I, Rusicke E, Aygören-Pürsün E, Heller C, Klingebiel T, Kreuz W. Characterization of acute hereditary angioedema attacks during pregnancy and breast-feeding and their treatment with C1 inhibitor concentrate. Am J Obstet Gynecol. 2010;203:131.e1-7.
164. Bouillet L, Longhurst H, Boccon-Gibod I, Bork K, Bucher C, Bygum A, et al. Disease expression in women with hereditary angioedema. Am J Obstet Gynecol. 2008;199:484.e1-4.
165. Czaller I, Visy B, Csuka D, Fust G, Toth F, Farkas H. The natural history of hereditary angioedema and the impact of treatment with human C1-inhibitor concentrate during pregnancy: a long-term survey. Eur J Obstet Gynecol Reprod Biol. 2010;152:44-9.
166. Bork K, Gul D, Dewald G. Hereditary angio-oedema with normal C1 inhibitor in a family with affected women and men. Br J Dermatol. 2006;154:542-5.
167. Bouillet L. Hereditary angioedema in women. Allergy Asthma Clin Immunol. 2010;6:17.
168. Baker JW, Craig TJ, Riedl MA, Banerji A, Fitts D, Kalfus IN, et al. Nanofiltered C1 esterase inhibitor (human) for hereditary angioedema attacks in pregnant women. Allergy Asthma Proc. 2013;34:162-9.
169. Chan W, Berlin N, Sussman GL. Management of hereditary angioedema with C1-INHibitor concentrate during two successive pregnancies. Int J Gynaecol Obstet. 2013;120:189-90.
170. Zuraw BL, Bork K, Binkley KE, Banerji A, Christiansen SC, Castaldo A, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33 Suppl 1:S145-56.
171. Bouillet L, Gompel A. Hereditary angioedema in women: specific challenges. Immunol Allergy Clin North Am. 2013;33:505-11.
172. Chinniah N, Katelaris CH. Hereditary angioedema and pregnancy Aust N Z J Obstet Gynaecol. 2009;49:2-5.
173. Prior N, Remor E, Gomez-Traseira C, Lopez-Serrano C, Cabanas R, Contreras J, et al. Development of a disease-specific quality of life questionnaire for adult patients with hereditary angioedema due to C1 inhibitor deficiency (HAE-QoL): Spanish multi-centre research project. Health Qual Life Outcomes. 2012;10:82.
174. Nordenfelt P, Dawson S, Wahlgren C-F, Lindfors A, Mallbris L, Bjorkander J. Quantifying the burden of disease and perceived health state in patients with hereditary angioedema in Sweden. Allergy Asthma Proc. 2014;35:185-90.
175. Bygum A, Andersen KE, Mikkelsen CS. Self-administration of intravenous C1-INHibitor therapy for hereditary angioedema and associated quality of life benefits. Eur J Dermatol. 2009;19:147-51.
176. Grumach AS, Valle SOR, Toledo E, de Moraes Vasconcelos D, Villela MMS, Mansour E, et al. Hereditary angioedema: first report of the Brazilian registry and challenges. J Eur Acad Dermatol Venereol. 2013;27:e338-44.
177. Bork K, Hardt J. Hereditary angioedema: long-term treatment with one or more injections of C1 inhibitor concentrate per week. Int Arch Allergy Immunol. 2011;154:81-8.
178. Cicardi M, Suffritti C, Perego F, Caccia S. Novelties in the Diagnosis and Treatment of Angioedema. J Investig Allergol Clin Immunol. 2016;26:212-21.
179. Riedl MA. The therapeutic landscape for hereditary angioedema: more change on the horizon? Ann Allergy Asthma Immunol. 2014;113:337-8.
180. Aygören-Pürsün E, Magerl M, Graff J, Martinez-Saguer I, Kreuz W, Longhurst H, et al. Prophylaxis of hereditary angioedema attacks: A randomized trial of oral plasma kallikrein inhibition with avoralstat. J Allergy Clin Immunol. 2016;138:934-5.
181. Chyung Y, Vince B, Iarrobino R, Sexton D, Kenniston J, Faucette R, et al. A phase 1 study investigating DX-2930 in healthy subjects. Ann Allergy Asthma Immunol. 2014;113:460-2.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888