Número Atual: Janeiro-Março 2023 - Volume 7 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Hemofagocitose em cultura de sangue periférico precedendo hemofagocitose em biópsia de medula óssea na síndrome hemofagocítica secundária
Hemophagocytosis in peripheral blood culture preceding hemophagocytosis in bone marrow biopsy in secondary hemophagocytic syndrome
Bruno Macedo Pinto1; Cristina Han Hue Lin2; Tainá Mosca1; Ademir Veras da Silva2; Gustavo Nunes Wakim2; Wilma Carvalho Neves Forte1
1. Faculdade de Ciências Médicas da Santa Casa de São Paulo, Disciplina de Imunologia - São Paulo, SP, Brasil
2. Irmandade de Misericórdia da Santa Casa de São Paulo, Hospital Central - São Paulo, SP, Brasil
Endereço para correspondência:
Wilma Carvalho Neves Forte
E-mail: wilmanevesforte@yahoo.com.br
Submetido em: 08/12/2022
Aceito em: 04/02/2023
Não foram declarados conflitos de interesse associados à publicação deste artigo
RESUMO
A síndrome hemofagocítica é determinada por desregulação do sistema imunológico, caracterizada por ativação excessiva de macrófagos, resultando em fagocitose de células sanguíneas normais no fígado, baço e medula óssea. Pode ser primária (genética) ou secundária (adquirida). Em adultos quase sempre é secundária, tendo infecções, neoplasias e doenças autoimunes como frequentes desencadeadores. Entre as principais manifestações da síndrome estão febre prolongada e hepatoesplenomegalia. O diagnóstico até o momento é confirmado pelo achado de hemofagocitose em biópsia de medula óssea. Entretanto, é descrito que a biópsia de medula óssea é normal nos primeiros dias de manifestações da síndrome. O presente relato tem como objetivo mostrar a observação de hemofagocitose em cultura de células de sangue periférico de paciente de 29 anos precedendo a hemofagocitose em biópsia de medula óssea. A paciente apresentava diferentes infecções, com grave comprometimento do estado geral e sem melhora com o tratamento das infecções. O achado laboratorial permitiu o tratamento precoce da síndrome hemofagocítica e a melhora da paciente. No presente relato a técnica utilizada está descrita detalhadamente para que possa ser reproduzida, além de ser apresentada uma revisão não sistemática da literatura sobre a síndrome.
Descritores: Linfo-histiocitose hemofagocítica, hiperferritinemia, infecção persistente.
Introdução
A síndrome hemofagocítica ou linfo-histiocitose hemofagocítica (HLH) ou síndrome de ativação macrofágica é determinada por aumento de citocinas pró-inflamatórias, com ativação excessiva de macrófagos, resultando em fagocitose de células sanguíneas normais no fígado, baço e medula óssea.
As principais manifestações são: febre (geralmente prolongada e acima de 38,5 °C) e hepatoesplenomegalia. Podem estar presentes manifestações: gerais (mal estar, astenia); hematológicas (coagulopatia, petéquias, púrpura, equimose, epistaxe); hepáticas; esplênicas; gastrointestinais (hematêmese, ascite, diarreia, vômitos, dor abdominal, hemorragia digestiva baixa); cutâneas (erupção eritematosa, edema, nódulos subcutâneos); respiratórias (tosse, dispneia, insuficiência respiratória); geniturinárias (síndrome nefrótica, insuficiência renal); distúrbios do sistema nervoso central (estado mental alterado, convulsões, encefalopatia, coma); e transtornos psiquiátricos (alterações de humor, delírio e psicose)1‑3. É necessária a investigação de HLH em casos de citopenia febril, pois o retardo do diagnóstico leva à disfunção de múltiplos órgãos e maior mortalidade.
As manifestações laboratoriais mais frequentes da HLH são: citopenias (anemia, trombocitopenia, neutropenia); aumento de ferritina e de triglicérides; diminuição de fibrinogênio; hemofagocitose tecidual; diminuição da função de células T e NK; aumento do receptor solúvel da interleucina-2 (sIL-2R ou CD25)2,3. Outras alterações também podem ser observadas, como aumento dos testes inflamatórios (PCR e VHS); aumento da lactato-desidrogenase; pleocitose do líquido cefalorraquidiano; aumento de transaminases hepáticas, bilirrubina total, ureia, creatinina; amônia normal ou levemente elevada; hipoalbuminemia; hiponatremia2,3.
As causas de HLH podem ser primárias ou secundárias, ambas com alta mortalidade. A HLH primária está relacionada a mutações genéticas em PRF1, UNC13D, STX11, STXBP2, SH2D1, XIAP, RAB27A, LYST, AP3B1, NLRC4, MAGTT1, ITK ou CD27, sendo mais frequente em crianças até 18 meses, mas pode eventualmente ocorrer em qualquer idade. A incidência da primária varia de 1 para 50.000 a 1.000.000 nascidos vivos, sem predomínio de sexo3. O perfil etiológico de HLH secundária varia muito em diferentes locais, sendo as etiologias infecciosas mais comuns nos Estados Unidos, França, Espanha e Coreia do Sul, e as neoplásicas mais frequentes na Itália, China, Japão e Taiwan1. Na América do Sul existem poucos casos descritos de HLH.
Habitualmente células TCD8 e NK liberam perfurinas (formam poros na superfície da célula-alvo) e promovem a ativação de granzimas (desencadeiam a apoptose)3,4. Para tanto, perfurinas e granzimas precisam ser sintetizadas, transportadas, englobadas em grânulos citolíticos e secretadas por TCD8 e NK. Mutações em genes que transcrevem proteínas envolvidas nestes quatro processos causam síndrome hemofagocítica primária, por incapacidade de interromper o estímulo antigênico, resposta imunológica permanentemente ativada, e aumento persistente das citocinas pró-inflamatórias TNF-α, IFN-γ, IL-1β, IL-2, IL-6, IL-12, IL-16, IL-183. Há também aumento da resposta anti-inflamatória, com aumento de IL‑10; entretanto, esta resposta compensatória não é suficiente para interromper a hiperativação macrofágica3. O microambiente de aumento de citocinas pró-inflamatórias resulta em ativação persistente de macrófagos, causando hemofagocitose, lesão tecidual, falência de órgãos e outras manifestações de HLH3,4.
A hipótese diagnóstica de HLH é feita por pontuações, segundo escores: HScore ou HLH-2004 (utilizado no presente relato) - ambos utilizados em adultos, tendo a mesma sensibilidade e especificidade; e HScore para crianças - HLH-20043,4.
O tratamento baseia-se em controlar o excesso da resposta imunológica, utilizando-se potentes imunossupressores, imunoglobulina humana, corticosteroides em altas doses e plasmaférese, além do tratamento da doença de base e dos danos multiorgânicos envolvidos.
O presente relato tem como objetivo mostrar a observação de hemofagocitose em cultura de células de sangue periférico de paciente de 29 anos precedendo a hemofagocitose em biópsia de medula óssea, e descrever detalhadamente a técnica utilizada para a observação da hemofagocitose em sangue periférico.
Relato de caso
Paciente do sexo feminino, 29 anos, natural da Bahia, procedente de São Paulo, Brasil, trabalhadora de coleta de lixo para reciclagem, portadora de infecção por HIV (há três anos havia abandonado o tratamento) e doença renal crônica (glomeruloesclerose segmentar e focal colapsante) por HIV. Procurou serviço de emergência por falta de ar, cansaço, e tosse produtiva há cinco semanas, com piora da dispneia há um dia. Foi internada e apresentou duas crises convulsivas, controladas com fenitoína. Apresentava-se consciente e orientada em tempo e espaço, hepatomegalia (3 cm), percussão com macicez no espaço de Traube, linfonodos inguinais indolores, palpáveis (2 cm) bilateralmente, sem sinais flogísticos, saturação de 96% em uso de cateter de oxigênio, taquicardia (135 bpm). Durante os seis meses de internação subsequentes apresentou febre diária de origem indeterminada, herpes cutâneo crônico em região perineal, infecção por Citomegalovirus, candidemia, tuberculose pulmonar e miocardite viral. No sexto mês de internação a paciente apresentou importante deterioração clínica, com quadro de síndrome da resposta inflamatória sistêmica. Os exames laboratoriais mostraram culturas negativas para possível bacteremia, pancitopenia, TCD4 11 células/mm3, hipertrigliceridemia e hiperferritinemia. Diante do quadro clínico e laboratorial foi aventada a hipótese de síndrome hemofagocítica. O Hscore for Reactive Hemophagocytic Syndrome5 indicou 96% de chance de HLH. Tendo em vista a hipótese de síndrome hemofagocítica, foi realizada biópsia de medula óssea, com resultado histopatológico normal. Foi então realizada cultura de fagócitos do sangue periférico, mostrando hemofagocitose em sangue periférico (Figura 1), iniciando-se tratamento para HLH. Três dias depois da cultura de sangue mostrar hemofagocitose, nova biópsia de medula óssea foi realizada, agora revelando síndrome hemofagocítica.
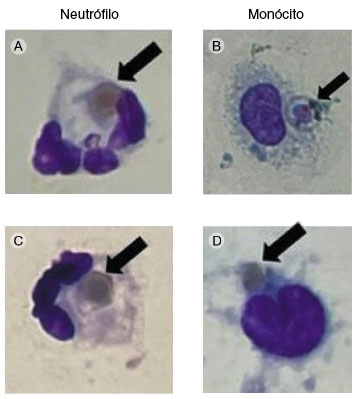
Figura 1
Cultura de células do sangue periférico mostrando hemofagocitose por neutrófilos e por monócitos. As setas indicam hemácias fagocitadas por neutrófilos (fotos A e C) e por monócitos (fotos B e D) de sangue periférico
Após o diagnóstico pela cultura de células, devido à indisponibilidade de outras medicações no hospital de atendimento e diante da piora rápida e progressiva da paciente foi realizada pulsoterapia com metilprednisolona (1 g EV por três dias). Após três dias de tratamento com corticosteroide a paciente apresentou remissão do quadro febril, melhora da dispneia, tornando-se confortável em ar ambiente, sem necessidade de suplementação com oxigênio; a frequência cardíaca caiu de 135 para 80-90 bpm. Após uma semana, a análise laboratorial mostrou diminuição da ferritina (2756 para 1075 ng/mL) e melhora da anemia (6,1 para 11,9 g/dL). Depois de quatro semanas a paciente continuou com melhora do estado geral, permanecendo afebril, eupneica, com frequência cardíaca normal, ganhando 5 kg de peso corporal, os exames laboratoriais hematológicos e as provas inflamatórias se normalizaram. Assim, a paciente recebeu alta hospitalar após a resolução completa das manifestações de HLH.
Discussão
A paciente apresentava febre alta e prolongada, hepatoesplenomegalia, sem sinais característicos de determinada infecção, além de resposta insatisfatória aos medicamentos. Apresentava ainda fatores de risco para desenvolver a síndrome hemofagocítica secundária: história de HIV, tuberculose prévia e tratamento para citomegalovírus ao longo da internação1,4,5. Tais fatores, associados às manifestações clínicas apresentadas pela paciente (febre alta e persistente, hepatoesplenomegalia, adenomegalia, pancitopenia e culturas negativas) foi feita a hipótese de HLH1,2. Tal doença, embora rara, tem uma maior incidência em portadores de HIV e de tuberculose, com pior prognóstico em pacientes com infecção ativa5.
A HLH secundária pode ser dividida em dois subgrupos: com desencadeantes infecciosos (virais, bacterianos, parasitários, fúngicos) e não infecciosos (neoplasias, doenças autoimunes, medicamentos e outras etiologias). Um terço de HLH secundária apresenta mais de uma causa1,4. A incidência da HLH secundária é de 1:800.000 indivíduos, predominando em adultos, com média de idade de diagnóstico aos 50 anos, e é mais prevalente no sexo feminino1.
A paciente apresentava diferentes causas infeciosas que poderiam desencadear HLH: HIV, tuberculose, Citomegalovirus. As causas infecciosas mais frequentes de HLH são: infecções por HIV e por Epstein-Barr virus1,4. Entre outros desencadeantes virais encontram-se: herpes vírus, Citomegalovirus, vírus de hepatite A,B,C, Parvovirus B19, Influenza virus e mais recentemente infecção por SARS-CoV-26. São referidas também infeções bacterianas (Mycobacterium tuberculosis, S. aureus, E. coli e Rickettsia spp.), parasitárias (Leishmania spp., Plasmodium spp., Toxoplasma spp.) e fúngicas (Histoplasma spp.)1,3-5,7.
As causas não infecciosas de HLH devem ser afastadas pelo quadro clínico ou laboratorial, como no presente caso. Os principais desencadeantes não infecciosos são: neoplasias (especialmente hematológicas, como linfomas, leucemias e tumores sólidos); doenças autoimunes (lúpus eritematoso sistêmico, doença de Still, artrite reumatoide, vasculites e doenças inflamatórias intestinais); medicamentos (metotrexato, anti-inflamatórios não hormonais, anticonvulsivantes, imunossupressores, quimioterapia, sais de ouro, sulfasalazina); procedimentos médicos (cirurgias, biópsias, nutrição parenteral, hemodiálise, transplantes hepático e renal); outras doenças (diabetes, doenças hepáticas crônicas); e gestação1,3.
No processo de diagnóstico da HLH é ainda importante excluir outras causas de imunodesregulação sistêmica, como sepse, síndrome da resposta inflamatória sistêmica e síndrome de disfunção de múltiplos órgãos, que apresentam quadros clínicos semelhantes1‑3, geralmente com evolução mais rápida, diferente do presente relato.
A hipótese diagnóstica da paciente foi feita pelo critério HLH-2004: presença de febre, esplenomegalia, citopenias, hipofibrinogenemia, hipertrigliceridemia, ferritina acima de 500 µg/L, resultando em escore de 96% de chance de ter HLH. A hipótese diagnóstica de HLH é feita na presença de cinco dos oito critérios clínicos do escore HLH-2004: (1) febre > 38,5 °C; (2) esplenomegalia; (3) citopenia de duas ou mais séries: hemoglobina < 9 g/L, ou plaquetas < 100.000 cels./mm3, ou leucócitos < 3.000 cels./mm3, ou neutrófilos < 1.000 cels./mm3; (4) hipertrigliceridemia > 265 mg/dL e/ou hipofibrinogenemia < 150 mg/dL; (5) hemofagocitose em medula óssea, baço, fígado ou linfonodos; (6) atividade de células NK baixa ou ausente; (7) hiperferritinemia > 500 ng/mL; (8) CD25 solúvel ou sIL-2R > 2.400 U/mL8.
O diagnóstico de certeza é feito pela observação de hemofagocitose em biópsia de medula óssea. Entretanto, tal biópsia costuma ser normal nos primeiros dias das manifestações clínicas da síndrome, como aconteceu no presente caso, e mesmo a ausência de hemofagocitose na biópsia de medula óssea não exclui HLH8.
A cultura de células do sangue periférico mostrou presença de células sanguíneas ou fragmentos das mesmas no interior de fagócitos mononucleares e de neutrófilos. Assim, recebeu o diagnóstico de HLH antes mesmo do aparecimento de hemofagocitose na medula óssea, e na falta de exames não disponíveis, como atividade de células NK e dosagem de IL-2R11.
A cultura de fagócitos no presente estudo foi realizada a 37 °C, em atmosfera com 5% de CO2. Os fagócitos do sangue periférico foram isolados por sedimentação espontânea9. A técnica de sedimentação espontânea garante uma suspenção celular com cerca de 77% de neutrófilos10. As demais células presentes são, em ordem de quantidade: hemácias, eosinófilos, monócitos e linfócitos. Assim, a cultura realizada desta forma permite o contato direto entre os diferentes tipos celulares, possibilitando que hemácias sejam fagocitadas por outras células sanguíneas, como ocorre na síndrome hemofagocítica, possibilitando o diagnóstico de HLH no presente relato. A literatura mostra que a biópsia de medula óssea geralmente apresenta-se normal nos primeiros dias da síndrome.
O tratamento precoce de HLH é de fundamental importância para a sobrevida dos portadores. A HLH convencionalmente é tratada com etoposide, rituximabe, ruxolitinibe ou emapalumabe11‑13, podendo ser utilizado transplante de células-tronco hematopoiéticas em pacientes refratários ao tratamento. Devido à indisponibilidade das drogas de primeira linha e em virtude da piora do quadro clínico foi instituída terapêutica de segunda opção: indução de pulsoterapia com metilprednisolona. Mesmo não sendo a primeira linha de tratamento, o uso de corticosteroides e o tratamento das infecções desencadeantes permitiu o controle das manifestações clínicas e laboratoriais. Após o tratamento do estado hiperinflamatório, a paciente apresentou remissão completa do quadro febril, melhora da dispneia, tornando-se confortável em ar ambiente e melhora dos exames laboratoriais. Após quatro semanas houve regressão total do quadro, melhora do estado geral, permitindo que a paciente recebesse alta com acompanhamento ambulatorial.
Conclusão
A cultura de sangue periférico do presente relato mostrou síndrome hemofagocítica por fagócitos neutrofílicos e mononucleares precedendo a hemofagocitose apontada pela biópsia de medula óssea.
O presente caso foi apresentado para mostrar que a cultura de sangue periférico pode auxiliar nos casos de hipótese clínica de síndrome hemofagocítica que apresentem biópsia normal de medula óssea ao início do quadro.
Referências
1. Ramos-Casals M, Brito-Zerón P, López-Guillermo A, Khamashta MA, Bosch X. Adult Hemophagocytic Syndrome. Lancet. 2014;383(9927):1503-16.
2. Sen ES, Steward CG, Ramanan AV. Diagnosing Hemophagocytic Syndrome. Arch Dis Child. 2017;102(3):279-84.
3. Al-Samkari H, Berliner N. Hemophagocytic Lymphohistiocytosis. Annu Rev Pathol. 2018;13:27-49.
4. Canna SW, Marsh RA. Pediatric Hemophagocytic Lymphohistio-cytosis. Blood. 2020;16;135(16):1332-43.
5. Revière S, Galicier L, Coppo P, Marzac C, Aumont C, Lambotte, et al. Reactive Hemophagocytic Syndrome in adults: a retrospective analysis of 162 patients. Am J Med. 2014;127:1118-25.
6. England JT, Abdulla A, Biggs CM, Lee AYY, Hay KA, Hoiland RL, et al. Weathering the COVID-19 storm: Lessons from hematologic cytokine syndromes. Blood Rev. 2021;45:100707.
7. Lerolle N, Laanani M, Galicier L, Rivière S, Meynard JL, Azoulay E, et al. Factors associated with tuberculosis-associated Hemophagocytic Syndrome: a multicentre case-control study. Int J Tuberc Lung Dis. 2020;24(1):124-30.
8. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. Diagnostic and therapeutic guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr Blood Cancer. 2007;48:124-31.
9. Forte WC, Gonzales CC, Carignani S, Mimica I. Evaluation of polymorphonuclear neutrophils in moderate malnutrition. Rev Assoc Med Bras. 1999;45(2):147-51.
10. Mosca T, Forte WCN. Comparative efficiency and impact on the activity of blood neutrophils isolated by Percoll, Ficoll and spontaneous sedimentation methods. Immunol Invest. 2016;45(1):29-37.
11. Marsh RA, Haddad E. How i treat primary Hemophagocytic Lymphohistiocytosis. Br J Haematol. 2018;182(2):185-99.
12. Hu S, Bansal P, Lynch D, Rojas Hernandez CM, Dayao Z. Rituximab, etoposide, methylprednisolone, high-dose cytarabine, and cisplatin in the treatment of secondary Hemophagocytic Lymphohistiocytosis with classical Hodgkin Lymphoma: a case report and review of the literature. J Med Case Rep. 2016;10(1):365.
13. Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat Hemophagocytic Lymphohistiocytosis. Blood. 2011;118(15):4041-52.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888