Número Atual: Janeiro-Março 2022 - Volume 6 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicações Clínicas e Experimentais
Tuberculose intratorácica na forma pseudotumoral e óssea como manifestação de doença granulomatosa crônica
Intrathoracic tuberculosis in the pseudotumoral and bone form as a manifestation of chronic granulomatous disease
Priscilla Filippo A. M. Santos1; Antonio Condino-Neto2; Lillian Nunes Gomes2; Claudete Araújo Cardoso3
DOI: 10.5935/2526-5393.20220014
1. Hospital Municipal Jesus, Ambulatório de Especialidades - Rio de Janeiro, RJ, Brasil
2. Instituto de Ciências Biomédicas da Universidade de São Paulo, Departamento de Imunologia - São Paulo, SP, Brasil
3. Faculdade de Medicina da Universidade Federal Fluminense, Departamento Materno-Infantil - Niterói, RJ, Brasil
Endereço para correspondência:
Priscilla Filippo A. M. Santos
E-mail: priscillaalvim05@gmail.com
Submetido em: 19/08/2021
Aceito em: 27/11/2021
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
A doença granulomatosa crônica (DGC) é um erro inato da imunidade de fagócitos, e ocorre em decorrência de mutações que afetam componentes da enzima NADPH oxidase. Os pacientes são suceptíveis a infecções graves e letais por fungos e bactérias. O objetivo deste trabalho é relatar o caso de um lactente com DGC que apresentou manifestação clínica de tuberculose (TB) intratorácica na forma pseudotumoral e óssea iniciada no período neonatal. O diagnóstico de DGC foi realizado através do teste de DHR e, após o início da profilaxia com sulfametoxazol-trimetroprima e itraconazol, o paciente manteve-se estável clinicamente. A mãe e a irmã também apresentaram DHR alterados, a análise genética revelou uma mutação ligada ao X no exon 2 do gene CYBB c.58G>A, levando uma alteração em G20R. É fundamental que o diagnóstico seja realizado o mais precocemente possível, a fim de instituir as orientações aos familiares e tratamento adequado, reduzindo assim complicações infecciosas e melhorando prognóstico.
Descritores: Doença granulomatosa crônica; doenças da imunodeficiência primária; tuberculose.
INTRODUÇÃO
A doença granulomatosa crônica (DGC) é uma doença genética heterogênea, que foi descrita pela primeira vez na década de 1950. É um erro inato da imunidade raro, caracterizado por infecções graves e recorrentes, devido ao comprometimento funcional do complexo NADPH oxidase em monócitos e granulócitos neutrofílicos1,2. Esse complexo enzimático gera superóxido e é essencial para a morte intracelular de patógenos, fungos e bactérias, pelos fagócitos. Além de apresentar efeitos citotóxicos diretos, a produção de espécies reativas de oxigênio parece ter importância para outras funções imunes inatas. A incidência é variável: 1:1 milhão na Itália2, 1:300.00 no Japão4, 1:250.000 nos Estados Unidos1 e 1: 70.000 na população árabe israelense5.
O pulmão, a pele, os linfonodos e o fígado são os órgãos mais acometidos1. Pacientes com DGC podem apresentar tuberculose (TB), que é uma infecção oportunista e de alta prevalência mundial, especialmente em países tropicais6,7. O desenvolvimento da doença pelo bacilo da TB depende da interação entre os fatores imunológicos do hospedeiro e a agressividade do agente infeccioso. A TB na forma pseudotumoral é uma entidade rara, sendo difícil o diagnóstico, podendo ser confundido com neoplasia primária ou secundária. As amostras bacteriológicas costumam ser negativas, o que pode causar um atraso no diagnóstico e na terapêutica6. O diagnóstico de DGC pode ser realizado pelo teste da dihidrorodamina (DHR), que mede a digestão intracelular de fagócitos, e é confirmado por estudos de genotipagem8.
Estudos recentes relataram uma taxa de sobrevida de aproximadamente 90% aos 10 anos de idade, atribuída ao diagnóstico precoce e instituição de antibioticoprofilaxia, uso de interferon-gama, profilaxia anti-fúngica e transplante de células-tronco hematopoiéticas9. Entretanto, a idade média da morte desses pacientes permanece em torno de 30 a 40 anos10.
O objetivo desse estudo é relatar um caso de TB intratorácica na forma pseudotumoral e óssea que é incomum, no período neonatal, como primeira manifestação de doença granulomatosa crônica. O projeto foi aprovado pelo Comitê de Ética em Pesquisa (CAAE: 46255021.4.0000.5259), sendo assinado um termo de consentimento livre e esclarecido pela responsável do paciente.
RELATO DE CASO
Menino, 12 meses, foi encaminhado para investigação imunológica com história de tuberculose (TB) intratorácica, na forma pseudotumoral e óssea (Figura 1). Recebeu a vacina BCG no período neonatal, sem complicações. Aos 19 dias de vida internou com quadro de febre diária, edema e eritema de primeiro quirodáctilo esquerdo. Durante a investigação da criança não se obteve confirmação microbiológica do Mycobacterium tuberculosis, mas pelo aspecto radiológico da imagem pulmonar fez-se o diagnóstico de TB, sendo na ocasião iniciados isoniazida, rifampicina e pirazinamida, com regressão do pseudotumor e remissão completa do quadro ósseo após 12 meses de tratamento. Os pais e a irmã foram investigados para TB, com radiografia de tórax normal e prova tuberculínica reatora, sendo prescrito na ocasião tratamento para infecção latente da tuberculose (ILTB). Os marcadores tumorais foram negativos. Posteriormente, o paciente foi internado duas vezes por pneumonia, sem necessidade de oxigenoterapia, com PCR positivo para SARS-CoV2 na segunda internação. O crescimento e desenvolvimento eram adequados para a idade. Não havia história familiar de erros inatos da imunidade, de abortos espontâneos recorrentes e de consanguinidade entre os pais. Apresentava cicatriz de BCG acima de 3 mm ao exame físico, sem outras alterações.
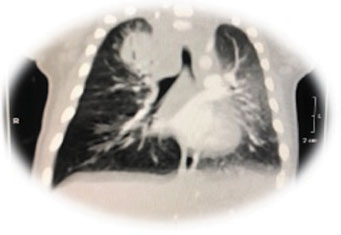
Figura 1
Angiotomografia de tórax - tuberculose intratorácica forma pseudotumoral
As sorologias para HIV I e II, HTLV I e II resultaram negativas. Hemograma evidenciou anemia. Dosagem de glicose-6-fosfato desidrogenase, imunoglobulinas, perfil linfocitário com resultados normais. Sorologias para sarampo e caxumba IgG reativas. No entanto, o ensaio de dihidrorodamina (DHR) apresentou resultado anormal da produção de ROS em granulócitos após a estimulação e foi sugestivo de DGC (Figura 2). Iniciada profilaxia com sulfametoxazol-trimetroprima e itraconazol com boa evolução clínica. Solicitada a tipagem de HLA e indicado transplante de células tronco hematopoiéticas. A mãe e a irmã apresentaram DHR alterados, sugestivos para portadoras de DGC ligada ao X (Figura 2).
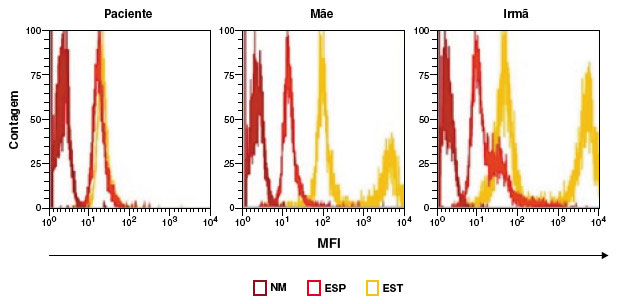
Figura 2
Resultado do teste de DHR. DHR de granulócitos espontâneo (ESP) ou estimulado com PMA (EST) e o não marcado, sem DHR (NM), de amostras do paciente, mãe e irmã, respectivamente, com resultado alterado do paciente, e sugestivo para portador de DGC ligado ao X
Além disso, foi realizado o sequenciamento do gene CYBB para confirmar o diagnóstico de DGC. Os dados da sequência foram analisados usando os bancos de dados NCBI (www.ncbi.nlm.nih.gov), Ensembl SNP (https://www.ensembl.org/index.html), e o Mutation Taster (http://www.mutationtaster.org). Esta análise confirmou uma mutação missense (c.58G>A) no exon 2 que leva a uma substituição de um único aminoácido, G20R na proteína gp91phox. A mesma mutação foi observada em heterozigose na mãe e irmã do paciente (Figura 3).
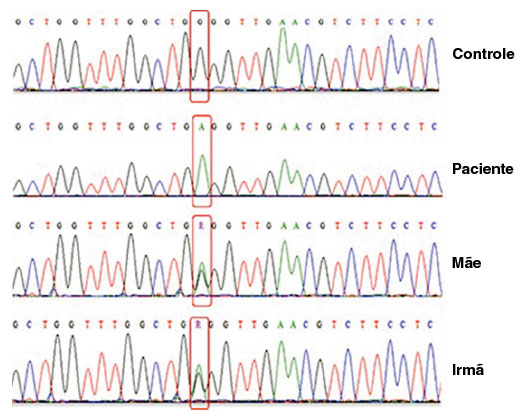
Figura 3
Resultados de sequenciamento para gene CYBB. Cromatograma para o Controle saudável, paciente, sua mãe e sua irmã, de cima para baixo, respectivamente. Mostra em destaque vermelho a substituição de um único nucleotídeo, c.58G> A, que resulta em uma mudança G20R
DISCUSSÃO
A DGC é um erro inato da imunidade raro, com incidência variável de acordo com a etnia, mais prevalente no sexo masculino (2:1), pelo modelo predominante de transmissão genética (doença ligada ao X)2. O lactente do relato de caso é do sexo masculino, e provavelmente apresenta uma doença ligada ao X, uma vez que sua mãe e irmã apresentaram DHR alterados. Entretanto, defeitos em genes autossômicos também podem causar DGC em homens e mulheres11. A DGC ligada ao X é mais comum em áreas com miscigenação, enquanto a forma autossômica recessiva é mais encontrada em áreas com histórico de consanguinidade12. Não há relato de consanguinidade entre os pais. Crianças com DGC ligada ao X têm apresentação clínica mais grave: início precoce da doença, obstruções frequentes causadas por granulomas, infecções mais frequentes e maior taxa de mortalidade11. Pacientes com DCG apresentam expressão e função reduzidas de receptores Toll-like, receptores de complemento e receptores de quimiocinas que se correlacionam com a gravidade da doença. A causa mais comum da DGC é um defeito na CYBB gene (gp91 phox), localizado no braço curto do cromossoma X (Xp21.1-p11.4)2.
Um estudo italiano multicêntrico mostrou que as manifestações infecciosas mais comuns foram infecção pulmonar (pneumonia/abscesso em 50%), dermatite/abscesso subcutâneo (46%), linfadenite (45%), osteomielite e abscesso hepático (16%)3. Arnold et al. observaram que o pulmão, a pele, os linfonodos e o fígado são os órgãos mais acometidos1. A osteomielite é uma infecção importante na DCG e pode surgir da disseminação hematogênica de patógenos (S. aureus , Salmonella spp. , S. marcescens) ou invasão contígua do osso11. A susceptibilidade a infecções nesses pacientes surge do período neonatal até a idade adulta, e o resultado depende do reconhecimento imediato e da terapia para a infecção subjacente12.
No Brasil, uma série de 18 pacientes identificou como as manifestações mais comuns de DGC: linfadenopatia, hepatoesplenomegalia, pneumonia e abscessos. Um segundo estudo, com sete pacientes brasileiros, mostrou que a pneumonia foi a manifestação clínica mais frequente, seguida por infecções de pele, sinusite, otite e abscesso hepático13.
Na Europa e na América do Norte, os patógenos mais comuns são Aspergillus spp. , Staphylococcus aureus , Burkholderia cepacia , Serratia marcescens , Nocardia spp. e Salmonella1,3. Nesses locais, não é comum a infecção por micobactérias. A DGC é a imunodeficiência primária em que é mais comum a infecção fúngica invasiva, afetando os pulmões e a parede torácica1. Em países em desenvolvimento, o Bacilo de Calmette-Guerin (BCG) e o Mycobacterium tuberculosis são os patógenos mais importantes11. Há relatos de infecção por micobactérias após a BCG e por Mycobacterium tuberculosis na China, no Irã e na América Latina. Entretanto, os pacientes desenvolvem uma forma grave localizada e não disseminada pela BCG e TB pulmonar e não miliar11.
Em países como o Brasil, em que a tuberculose é endêmica, a vacinação com BCG é feita nos primeiros meses de vida, sendo usualmente aplicada no período neonatal. E os pacientes com imunodeficiência podem apresentar reações adversas à vacinação BCG. Um estudo multicêntrico (América Latina, África, Europa e Ásia) realizado por Conti F. e cols. mostrou que a reação adversa ao BCG foi o primeiro sinal da doença em 39 (55%) das 53 crianças com essa reação. Uma reação local ou regional (BCG-ites) foi relatada em 33 (63%) dos 53 pacientes com reação adversa, e uma reação disseminada (BCG-osis) foi observada nos outros 20 (37%) pacientes14. Além disso, os pacientes com qualquer forma de DGC podem falhar no desenvolvimento de uma resposta imune protetora contra espécies de Mycobacterium, e podem desenvolver TB ativa em qualquer fase da vida. Clinicamente, a infecção por BCG foi em alguns casos a primeira manifestação de DGC no Brasil14.
Na Argentina, Hong Kong e Iran, até 11%, 54,5% e 31,7% dos pacientes com DGC, respectivamente, apresentaram TB. Crianças com formas graves de tuberculose devem ser investigadas para erros inatos da imunidade14.
Em um estudo realizado por Oliveira Júnior e cols. com dados do LASID (Latin American Society for Immunodeficiencies), foram avaliados 71 pacientes com DCG com a seguinte distribuição por países: 39% do Brasil, 36% da Argentina, 16% do México, 6% do Chile e 3% da Colômbia. Dos pacientes avaliados, 30% apresentaram reação a BCG com uma prevalência aumentada no grupo com mutação no gene CYBB (85,7%), comparado ao grupo com mutação no gene NCF1 (14,3%). Destes pacientes que apresentaram reação à BCG, 30% apresentou reação disseminada com quadro clínico grave13.
Pacientes com DGC podem apresentar alteração na resposta inflamatória e autoimunidade1. As manifestações inflamatórias são comuns em pacientes com DGC e são observadas com mais frequência no trato gastrointestinal, trato urogenital, pulmões e olhos2.
Uma das características da DCG é a formação de granulomas, podendo causar sintomas clínicos de obstrução, como vômitos, disfagia, esvaziamento gástrico lento, perda de peso, obstrução brônquica, obstrução da bexiga, etc.11. No caso relatado, a primeira manifestação clínica foi TB intratorácica na forma pseudotumoral, que regrediu após o início do tratamento específico.
O Brasil está entre os 30 países com maior carga de TB no mundo, com uma taxa de incidência de 80.000 casos/ano. As crianças representam 10% dos casos totais de TB. As crianças menores de cinco anos de idade apresentam maior risco de adoecimento após a infecção primária, quando comparadas aos adultos e adolescentes. A forma pulmonar é a mais frequente, destacando-se o maior potencial de evolução para formas graves de TB, como a meningoencefalite e a miliar. A vacina BCG protege contra essas formas graves e no Brasil, sendo a sua aplicação recomendada ao nascimento. Pacientes com TB devem ser investigados para HIV15. As manifestações radiológicas mais frequentes na infância são: linfonodomegalias (hilares ou mediastinais), doença miliar (tipo reticulonodular difusa), doença parenquimatosa (condensação pulmonar), atelectasia e derrame pleural (raramente em menores de cinco anos). As alterações na tomografia computadorizada são as consolidações lobares e áreas hipodensas, encontradas em mais de 80% dos casos, e, em menos de 25%, as cavitações16. A angiotomografia computadorizada de tórax do paciente evidenciou massa mediastinal com centro hipodenso e pequena escavação no seu interior associado a adenopatia hilar, paratraqueal, infracarinal sugestivo de TB.
O diagnóstico de TB em 80% dos casos em crianças menores que 5 anos é feito sem a comprovação bacteriológica, devido à característica paucibacilar da doença na faixa etária pediátrica, o que dificulta a realização de bacterioscopia de escarro em crianças menores16. Na ocasião do diagnóstico de TB na criança, deve-se sempre pesquisar a história de doença nos contactantes, a fim de se identificar o caso fonte da TB. No caso relatado, os pais e a irmã não apresentaram a doença. O paciente não teve a comprovação bacteriológica, e apresentou regressão completa do quadro clínico com a instituição do tratamento com esquema RIP.
Marine B. e cols. relataram que o diagnóstico de DGC foi feito em média com 4,4 anos (mediana de 2,5 anos, faixa de 0 a 38 anos) e a média de idade do início dos sintomas foi de 1 ano (mediana 7,5 anos, faixa de 0 a 10 anos)13. O paciente iniciou os sintomas precocemente, no período neonatal e o diagnóstico de DGC foi realizado aos 13 meses, através do DHR que foi realizado e repetido após 15 dias. O DHR é um ensaio muito sensível e específico que detecta de forma confiável todas as deficiências de NADPH oxidase em neutrófilos18. O diagnóstico precoce é fundamental para a instituição adequada do tratamento, com uso de antibiótico e antifúngico profiláticos, evitando complicações infecciosas, internações e interferindo na qualidade de vida do paciente. Além disso, é importante encontrar a mutação genética, para aconselhamento genético e eventualmente escolher um doador de medula óssea adequado ou realizar terapia gênica10,17. No Brasil, ainda não temos disponível a terapia gênica.
Em alguns casos, os pais de paciente com DGC podem considerar ter mais um filho para servir como doador de células-tronco hematopoéticas para o irmão afetado12. Neste relato de caso, os pais, em princípio, não pensam em ter mais filhos. Pensando no aconselhamento genético, foi solicitado o DHR e análise genética materna e da irmã. Os resultados dos DHRs tiveram resultados alterados e a genética confirmou que ambas apresentam a mesma variante encontrada no paciente (c.58G>A), na forma heterozigota. Esta é uma mutação missense no exon 2 do gene CYBB, resultando na mudança de aminoácido p.G20R no domínio N-terminal. Essa é uma mutação já descrita18. Os dados deste estudo destacam a relevância do diagnóstico genético para o aconselhamento familiar definitivo, visto que uma nova gestação da mãe tem 50% de chance de nascer um menino com DGC, o mesmo para a futuras gestações da irmã.
O uso profilático com sulfametoxazol-trimetoprima ou doxicacilcina e itraconazol a longo prazo mostrou a redução de infecções em pacientes com DGC, e é recomendado durante os períodos sem infecção19,20. A partir do início da profilaxia com essas medicações, o paciente ficou clinicamente estável, sem outras internações devido a intercorrências infecciosas. Recomenda-se também a prevenção de infecções por meio de imunizações11.
A BCG é uma vacina obrigatória na América Latina. É geralmente administrada durante o período neonatal. Esta prática é potencialmente prejudicial para os pacientes com DCG, SCID ou outra imunodeficiência que afeta os fagócitos ou função da célula T. Testes de triagem neonatal para DCG e outros defeitos de fagócitos deveriam ser realizados antes da vacinação BCG13.
No México e na Argentina, os pacientes com DGC têm acesso à terapia de rotina com interferon-gama, feita por via subcutânea, três vezes por semana. No Brasil, esse medicamento não é registrado pela agência nacional reguladora, limitando o acesso para uso13.
No estudo de Oliveira-Junior e cols. foram realizados transplantes de medula óssea em pacientes no Brasil, México e Argentina, dependendo da disponibilidade de um doador compatível. A terapia gênica para imunodeficiências primárias ainda não está disponível na América Latina13.
Agudelo-Flórez e cols. descreveram uma série de 14 pacientes com DCG da América Latina (10 brasileiros, 2 chilenos e 2 mexicanos), em que todos iniciaram quadro infeccioso antes de 2 anos. Nenhum apresentou reação grave à BCG. Todos fizeram uso profilático de sulfametoxazol/trimetoprim, 11 fizeram itraconazol e 2 receberam interferon gama recombinante regularmente21.
Outros diagnósticos diferenciais devem ser excluídos, como a deficiência de G6PD, porque essa deficiência na forma grave pode levar à formação insuficiente de NADPH nos leucócitos, dificultando a atividade da NADPH oxidase11.
Martire B. e cols. relataram uma taxa de sobrevida de 97%, 83% e 46% aos 10, 20 e 25 anos respectivamente, desde o diagnóstico da doença3.
Relatamos um caso raro de lactente com forma pseudotumoral e óssea de TB, sendo importante afastar outras causas com clínica e imagens semelhantes e investigar erros inatos da imunidade nesses casos, o que é relevante para o tratamento precoce e aconselhamento genético.
AGRADECIMENTOS
Agradecemos ao paciente e aos seus familiares por sua cooperação no estudo.
REFERÊNCIAS
1. Arnold DE, Heimall JR. A Review of Chronic Granulomatous Disease. Adv Ther. 2017;34(12):2543-57.
2. Rider NL, Jameson MB, Creech CB. Chronic Granulomatous Disease: Epidemiology, Pathophysiology, and Genetic Basis of Disease. J Pediatric Infect Dis Soc. 2018;7(Suppl 1):S2-S5.
3. Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T, Finocchi A, et al.; IPINET. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. 200;126(2):155-64.
4. M. Hasui. Chronic granulomatous disease in Japan: incidence and natural history. The study group of Phagocyte Disorders of Japan. Pediatr Int. 1999;41:589-93.
5. Wolach B, Gavrieli R, de Boer M, van Leeuwen K, Berger-Achituv S, Stauber T, et al. Chronic granulomatous disease: Clinical, functional, molecular, and genetic studies. The Israeli experience with 84 patients. Am J Hematol. 2017;92(1):28-36.
6. Snene H, Ben Mansour A, Toujani S, Ben Salah N, Mjid M, Ouahchi Y, et al. La tuberculose pseudotumorale, un diagnostic difficile [Tuberculous pseudotumour, a challenging diagnosis]. Rev Mal Respir. 2018 Mar;35(3):295-304. French. doi: 10.1016/j.rmr.2017.03.038. Epub 2018 Apr 5.
7. Silva GA, Brandão DF, Vianna EO, Sá Filho JBC, Baddini-Martinez J. Cryptococcosis, silicosis, and tuberculous pseudotumor in the same pulmonary lobe. J Bras Pneumol. 2013;39(5):620-6.
8. Yu JE, Azar AE, Chong HJ, Jongco III AM, Prince BT. Considerations in the Diagnosis of Chronic Granulomatous Disease. Journal of the Pediatric Infectious Diseases Society, 2018;v7(Suppl 1):6-11.
9. Kuhns DB, Alvord WG, Heller T, et al. Residual NADPH oxidase and survival in chronic granulomatous disease. N Engl J Med. 2010;363(27):2600-2610.
10. Jones LB, McGrogan P, Flood TJ, Gennery AR, Morton L, Thrasher A, et al. Special article: chronic granulomatous disease in the United Kingdom and Ireland: a comprehensive national patient-based registry. Clin Exp Immunol. 2008;152(2):211-8.
11. Roos D. Chronic granulomatous disease. Br Med Bull. 2016;118(1):50-63.
12. Chiriaco M, Salfa I, Di Matteo G, Rossi P, Finocchi A. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr Allergy Immunol. 2016;27(3):242-253.
13. de Oliveira-Junior EB, Zurro NB, Prando C, Cabral-Marques O, Pereira PV, Schimke LF, et al. Clinical and Genotypic Spectrum of Chronic Granulomatous Disease in 71 Latin American Patients: First Report from the LASID Registry. Pediatr Blood Cancer. 2015;62(12):2101-7.
14. Conti F, Lugo-Reyes SO, Blancas Galicia L, He J, Aksu G, Borges de Oliveira E Jr, et al. Mycobacterial disease in patients with chronic granulomatous disease: A retrospective analysis of 71 cases. Allergy Clin Immunol. 2016;138(1):241-248.e3.
15. Garest CZ, Campos IA, Mello SR, Barbosa AP, Araújo PA, Sias SA, et al. A Tuberculose pulmonar, em lactente jovem, diagnosticada pela avaliação de contato - Relato de caso. Resid Pediatr. 2021;11(1): - Relato de Caso. doi: 10.25060/residpediatr-2021.v11n1-117 .
16. Jardim VMJ, Alves JDC, Eduardo EFAF, Silva RS, Lima RA, Fonseca SCB. Tuberculose pulmonar confirmada em lactente: relato de caso. Resid Pediatr 2020;9(0). doi: 10.25060/residpediatr-2021.v11n2-138.
17. Constantino TSS, Goudouris ES. Doença granulomatosa crônica: um relato de caso. Resid Pediatr. 2020;0(0).
18. Di Matteo G, Giordani L, Finocchi A, Ventura A, Chiriaco M, Blancato J, et al.; IPINET (Italian Network for Primary Immunodeficiencies). Molecular characterization of a large cohort of patients with Chronic Granulomatous Disease and identification of novel CYBB mutations: an Italian multicenter study. Mol Immunol. 2009;46(10):1935-41.
19. Segal BH, Leto TL, Gallin JI, Malech HL, Holland SM. Genetic, biochemical, and clinical features of chronic granulomatous disease. Medicine (Baltimore). 2000;79(3):170-200.
20. Lee PP, Chan KW, Jiang L, Chen T, Li C, Lee TL, et al. Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J. 2008;27(3):224-30.
21. Agudelo-Flórez P, Prando-Andrade CC, López JA, Costa-Carvalho BT, Quezada A, Espinosa FJ, et al. Chronic granulomatous disease in Latin American patients: clinical spectrum and molecular genetics. Pediatr Blood Cancer. 2006;46(2):243-52.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888