Número Atual: Julho-Setembro 2021 - Volume 5 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
O estado da arte das síndromes autoinflamatórias associadas à criopirina
The state of the art of cryopyrin-associated periodic syndromes
Katharina Ruth Pagotto Betzler1,2; Victor Chiosini3,4; Alex Isidório Prado2,4; Bruna Gehlen4; Fabio Fernandes Morato Castro4; Myrthes Toledo Barros4; Jorge Kalil4; Leonardo Oliveira Mendonça2,4
DOI: 10.5935/2526-5393.20210040
1. Faculdade de Medicina de Graz, Faculdade de Medicina - Graz, Graz, Áustria
2. Centro de Doenças Raras e da Imunidade, Doenças Raras e da Imunidade - São Paulo, SP, Brasil
3. Faculdade de Medicina do Rio Grande, FURG - Rio Grande, RS, Brasil
4. Disciplina de Imunologia Clínica e Alergia, Imunologia Clínica e Alergia, FMUSP - São Paulo, SP, Brasil
Endereço para correspondência:
Leonardo Oliveira Mendonça
E-mail: leonardo.oliveira.mendonca@gmail.com
Submetido em: 07/03/2021
Aceito em: 25/04/2021
Não foram declarados conflitos de interesse associados à publicação deste artigo
RESUMO
As síndromes autoinflamatórias associadas à criopirina (CAPS) compreendem um grupo espectral de doenças raras autoinflamatórias. Todas estas doenças estão relacionadas ao inflamassoma NLRP3, sendo que de 50-60% dos pacientes apresentam mutações ao longo do gene NLRP3. Clinicamente, febre recorrente associada à urticária neutrofílica e outros sintomas sistêmicos são o grande marco clínico, comum a todo o espectro. O bloqueio da interleucina-1 trouxe grande alívio ao tratamento destas desordens, mas variações na resposta clínica podem ser observadas, principalmente nos espectros mais graves. Neste trabalho os autores trazem uma revisão do estado da arte das doenças autoinflamatórias CAPS. Foi realizado levantamento de literatura e, ao final, 49 artigos restaram como base para construção do texto final. O trabalho traz de forma narrativa os principais pontos relacionados a imunofisiopatologia, manifestação clínica, diagnóstico, tratamento, complicações e novas armas diagnósticas, e terapia gênica.
Descritores: Doenças hereditárias autoinflamatórias, síndromes periódicas associadas à criopirina, doenças genéticas inatas.
INTRODUÇÃO
As síndromes associadas à criopirina (CAPS - do inglês Cryopirin Associated Periodic Syndrome) representam um grupo espectral de doenças associadas a mutações no inflamassoma NLRP3, que é codificado pelo gene do mesmo nome (MIM #606416)1. Ao menos três síndromes bem definidas podem ser enquadradas neste espectro, sendo elas: Síndrome Autoinflamatória Familiar Associada ao Frio (FCAS - Familial Cold Autoinflammatory Syndrome; OMIM #120100), Síndrome Muckle-Wells (SMW - OMIM #191900), e Síndrome CINCA/NOMID (CINCA - Chronic-Infantile-Neurological-Cutaneous-Articular; OMIM #607115)2. Apesar de tais síndromes serem didaticamente classificadas em três doenças distintas, na prática clínica existe um continuum entre elas. O principal marco clínico comum a todas é o rash urticariforme, de natureza neutrofílica, associado à febre recorrente e a outros sinais sistêmicos como mal estar, fadiga, dor abdominal e eritema ocular (conjuntivite), sempre acompanhados de elevação de provas inflamatórias. O que difere as três síndromes é o grau de acometimento neurológico e articular1,2. Curiosamente, dois outros genes também foram associados ao perfil clínico das FCAS, sendo eles (NLPR12; MIM #609648)3,4 e (NLRC4; MIM #606831)5.
Os inflamassomas são complexos multiproteicos localizados no citoplasma celular. Eles atuam como "alarmes" imunológicos que disparam diante de receptores de reconhecimento de padrões (PRRs), tanto de infecção quanto de perigo, desde que estejam dentro da célula. Os inflamassomas ativam caspases inflamatórias (cysteine-dependent aspartate-directed proteases) que promovem a maturação de duas potentes citocinas pró-inflamatórias, a interleucina 1 (IL-1) e a interleucina 18 (IL-18). Este processo culmina com um tipo peculiar de morte celular denominada piroptose6-8.
Nesse trabalho, os autores propõem-se a revisar o estado da arte dos múltiplos espectros biológicos, clínicos, genéticos e terapêuticos das CAPS.
MATERIAIS E MÉTODOS
Foram feitas buscas na rede de dados PubMed utilizando os termos NLRP3, NLRP12, NLRC4, CAPS, Muckle-Wells, e CINCA/NOMID, sem uso de qualquer filtro. Os seguintes temas, isolados ou em conjunto, foram considerados na seleção dos artigos: diagnóstico clínico, diagnóstico laboratorial, diagnóstico genético, tratamento clínico, transplante de medula óssea, terapia gênica e prognóstico. Os resultados foram descritos de forma narrativa com revisão crítica dos principais tópicos levantados.
RESULTADOS
Ao todo 123 artigos foram selecionados. Após exclusão de dados duplicados ou não relevantes associados ao tema, 49 restaram como base para a construção do manuscrito final.
Imunofisiopatologia
A montagem do inflamassoma no citoplasma celular depende inicialmente do estímulo. A maioria dos inflamassomas é composta por três unidades, sendo uma sensora, um componente adaptador e um componente efetor, como a caspase 1. As moléculas sensoras podem ser de duas classes, os receptores associados a NOD (NOD-like receptors, NLRs) ou receptores semelhantes ao AIM2 (AIM2-like receptores, ALRs). Apesar do genoma humano ser capaz de formar 22 receptores NLRs, somente NLRP1, NLRP3, NLRP6, NLRP7, NLRP12 e NLRC4 são formados nos seus respectivos inflamassomas. O sensor essencial é formado por um domínio recrutador de caspase (CARD), um complexo pirina (PYD), um domínio oligomerizante e ligador de nucleotídeos (NACHT) e um domínio repetitivo e rico em leucinas (LRR). Curiosamente, os domínios NACHT e LRR são essenciais para manterem o inflamassoma inativo. Para se montar, um ASC recruta via PYD e CARD a pro-caspase 19-12.
Um destes complexos inflamassomas e de relevância neste artigo é o NLRP3 (NOD, LRR, e domínio pirina contendo proteína 3) que detecta diversos resquícios microbianos, sinais de perigo endógenos e irritantes internos/externos. Após contato com qualquer um destes sensores ocorre formação e ativação do complexo tripartite (inflamassoma NLRP3). A montagem deste complexo leva à liberação de citocinas pró-inflamatórias dependentes da caspase 1, que é uma das três partes do inflamassoma NLRP3. As principais citocinas liberadas são: IL-1β, IL-18 e isso leva à morte celular por piroptose mediada por gasdermin-D, que forma poros na parede da célula. Mutações ao longo do gene NLRP3 culminam tanto com reconhecimento como ativação aberrante deste inflamassoma e, consequentemente, com sintomas sistêmicos mediados por tais citocinas e marcadores característicos das CAPS7,13.
Manifestações clínicas
A Figura 1 resume as principais síndromes clínicas associadas às CAPS.
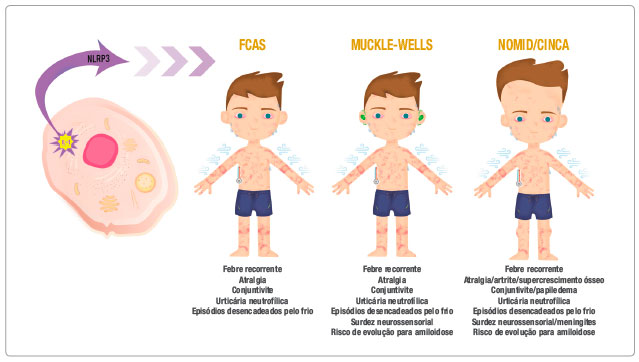
Figura 1 Desenho representativo da ativação do inflamassoma NLRP3, secreção de interleucina-1 e suas respectivas síndromes clínicas: FCAS, Síndrome de Muckle-Wells e Síndrome CINCA. Abaixo de cada síndrome estão as principais características clínicas atribuídas a elas
IL-1 = interleucina-1, FCAS (Familial Cold Autoinflammatory Syndrome) = Síndrome Autoinflamatória Familiar Associada ao Frio, CINCA (Chronic-Infantile-Neurological-Cutaneous-Articular) = Crônico-Infantil-Neurológica-Cutânea-Articular.
FCAS (FCAS - Familial Cold Autoinflammatory Syndrome; OMIM #120100)
A FCAS (síndrome familiar autoinflamatória induzida pelo frio) é o fenótipo menos severo dentre o espectro das síndromes associadas ao gene NLRP314. Ainda assim, a FCAS frequentemente tem grave impacto na qualidade de vida dos pacientes, de modo a impedir, por exemplo, que 95% dos sintomáticos façam atividades ao ar livre15.
Clinicamente, a doença é caracterizada por episódios recorrentes e autolimitados de febre, erupções cutâneas semelhantes à urticária e poliartralgia/artrite. Os episódios surgem geralmente na infância e são desencadeados pela exposição ao frio, permanecendo por 12-24h após o início do surto15. Sintomas adicionais menos frequentes podem ser: fadiga, cefaleia, conjuntivite e dores musculares, sobretudo em estações frias. Ainda, sintomas mais raros, mas que podem estar associados são: sudorese intensa, sede extrema, dor de cabeça e náusea, sempre que expostos ao frio16.
Casos de sobreposição pertencentes ao continuum são raramente reportados. Hedrick e cols. reportaram o caso de uma paciente com FCAS que apresentou episódios inflamatórios ao longo de vários dias, além de sintomas mais graves como artrite e miosite, geralmente associados à síndrome de Muckle-Wells16.
Síndrome de Muckle-Wells (SMW; OMIM #191900)
A Síndrome de Muckle-Wells representa a forma moderada/intermediária das criopirinopatias1. A perda auditiva neurossensorial e a amiloidose pertencem às manifestações mais graves de SMW devido à inflamação crônica, principalmente quando a doença não é tratada17-19. De forma geral, as manifestações clínicas parecem ser semelhantes ao quadro clínico de FCAS: febre recorrente, erupções cutâneas semelhantes à urticária (utricaria-like rash) e artrite14,16. Além disso, sintomas frequentes são conjuntivite, fadiga, e cefaleia1. Normalmente os sintomas de SMW surgem desde os primeiros dias até aos primeiros meses de vida20. Consequentemente, as manifestações clínicas como fadiga e cefaleia podem ter um impacto na vida do paciente e resultar em isolamento social21. A maioria dos pacientes com SMW apresenta crises mensais que duram de um a três dias1. O único sintoma osteoarticular relacionado à SMW é artrite/artralgia e até hoje nenhum caso com proliferação cartilaginosa foi reportado14,16,17.
Existem sobreposições clínicas no continuum entre MWS e NOMID. Normalmente, o envolvimento neurológico na síndrome de Muckle-Wells limita-se à surdez neurossensorial1. Além disso, a infertilidade é uma manifestação já descrita originalmente, relacionada à produção excessiva da interleucina 1 (IL-1), mas de mecanismos incertos. Em alguns pacientes observou-se oligospermia e azoospermia, provavelmente devido à inflamação crônica, aos níveis alterados de testosterona e à presença de amiloidose. O tratamento precoce pode ser benéfico para evitar a castração imunológica22; contudo, ainda é incerto se o bloqueio completo da IL-1 durante a infância ou adolescência poderá trazer problemas relacionados à maturação sexual14.
CINCA (CINCA - Chronic-Infantile-Neurological-Cutaneous-Articular; OMIM #607115)
A síndrome CINCA/NOMID representa o fenótipo mais grave no contexto do espectro clínico das CAPS23. Se não tratados precocemente, esses pacientes desenvolvem danos permanentes no sistema nervoso central devido à inflamação crônica24. A condição clínica mais grave observada na síndrome NOMID/CINCA é a meningite asséptica com aumento da pressão intracraniana, resultando a partir disso em hidrocefalia, atrofia cerebral e papiledema crônico. Cerca de 70% dos pacientes desenvolvem crescimento ósseo excessivo, resultando em contraturas articulares14,23. Crianças portadoras da síndrome usualmente apresentam manifestações cutâneas e articulares, como febre ao nascimento ou de forma recorrente nos primeiros anos de vida14.
A inflamação óssea e articular pode variar desde artralgias a artropatias graves, sendo isso determinado pela ossificação endocondral anômala e pelo crescimento ósseo. O envolvimento articular é geralmente assimétrico e afeta principalmente os joelhos. Outros achados musculoesqueléticos em pacientes com NOMID podem incluir raquitismo e osteomielite multifocal recorrente crônica25.
Em NOMID, as manifestações neurológicas são comuns. Tomografia computadorizada (TC) e ressonância magnética podem revelar leve proeminência dos ventrículos e aumento do líquido extra-axial, sugerindo atrofia cerebral leve, bem como calcificações da foice cerebral e da dura-máter, realce leptomeníngeo e inflamação coclear25,26. A síndrome é geralmente associada a prematuridade, retardo de crescimento intrauterino e oligoidrâmnio. Além dos sintomas típicos, Paccaud e cols. reportaram um paciente masculino com 33 semanas de vida, que apresentou-se com colestase, lesão endocárdica, polidrâmnio, anemia grave e trombocitopenia, tendo sido estes dois últimos achados associação bem descrita com CINCA27.
DIAGNÓSTICO E DIAGNÓSTICOS DIFERENCIAIS
Diagnóstico
O diagnóstico das síndromes CAPS é baseado em um tripé: clínico, laboratorial e genético. Clinicamente, os pacientes devem apresentar os sintomas já descritos acima, sendo que laboratorialmente, é comum o achado de anemia relacionada à doença crônica ao diagnóstico. Nas crises também são frequentes achados de leucocitose com neutrofilia, elevação de provas inflamatórias como proteina C-reativa (PCR), velocidade de hemossedimentação (VHS) e substância amiloide A (SAA). Nas FCAS, doenças de espectro mais leve, estes marcadores tendem à normalização nos períodos intercrises. Já nas síndromes mais graves, estes parâmetros tendem a estar constantemente elevados, mas com piora durante as crises28. Outro achado laboratorial importante é a presença de infiltrado neutrofílico na biópsia das lesões cutâneas, que se assemelham à urticária.
O critério diagnóstico proposto para CAPS e seus subtipos é baseado na presença de marcadores inflamatórios elevados (CRP ou SAA) somados a pelo menos dois tipos dos seis sinais e sintomas típicos de CAPS, sendo eles: (1) erupção cutânea semelhante à urticária, (2) episódios de inflamação desencadeados pelo frio, (3) perda auditiva, (4) artralgia ou artrite e mialgia, (5) meningite asséptica crônica, e (6) anormalidades esqueléticas, como crescimento epifisiário aumentado ou protuberância craniana frontal. Na presença dos referidos sinais e sintomas, não se faz necessária a demonstração da presença de mutação no gene NLRP329, embora haja na literatura o relato de que o critério diagnóstico não foi preenchido em 59% dos casos de uma família afetada com essa condição30.
Outros dados podem ajudar na identificação de síndromes especificas ou até mesmo de suas complicações. Em NOMID/CINCA, o achado radiográfico característico é a hipertrofia incomum da parte ossificada da cartilagem em crescimento, surgindo como uma massa calcificada heterogênea com deformidade associada a epífises e metáfises adjacentes25. Testes adicionais diagnósticos de complicações oculares e auditivas incluem fundoscopia, exames neurológicos e audiometria usando frequências de 500, 1.000, 2.000, 4.000, 6.000 e 8.000 Hz para a avaliação da condução de ar de tom puro31.
Diagnósticos diferenciais
Um dos principais diagnósticos diferenciais incluem outras doenças pertencentes ao grupo das Síndromes Periódicas Febris (SPF), tais como: Síndrome Autoinflamatória Associada ao Receptor de TNF (TRAPS), Síndrome Autoinflamatória Associada à Hiperglobulinemia D (Hiper IgD) e Febre Familiar do Mediterrâneo (FMF), dentre outras síndromes monogênicas ainda mais raras. Contudo, o diagnóstico diferencial baseado somente em manifestações clínicas é difícil por diversas razões, tais como: ausência de sinais patognomônicos nestas doenças; presença de sintomas que não se enquadram em nenhuma classificação; a maioria dos casos reportados são esporádicos, o que não corrobora para análises genéticas de segregação intrafamiliar; a maioria dos genes identificados nas SPF corresponde a variações "missense", cujos efeitos deletérios não estão bem elucidados32.
Outro diagnóstico importante a ser considerado em pacientes adultos é a síndrome de Schnitzler, uma síndrome autoinflamatória rara que pode ser confundida com CAPS, visto que o bloqueio de IL-1 tem importante papel no tratamento de ambas doenças e alguns pacientes carreiam variantes no mesmo gene, NLRP3. Outras síndromes não autoinflamatórias que também cursam com urticária devem ser consideradas, tais como: vasculite urticariforme, urticária ao frio, crioaglutininas, crioglobulinemias, sindrome de Wells e síndrome de Sweet, dentre outras raridades33.
Genética
A Síndrome Periódica Associada à Criopirina (CAPS) é uma doença genética de herança autossômica dominante decorrente de uma mutação genética do gene NLRP3, também chamado CIAS1 (Cold Induced Autoinflammatory Syndrome 1), NALP3 ou PYPAF134. Curiosamente, o fenótipo FCAS já foi associado a outros dois genes o NLRP12 e o NLRC43,5. Em relação ao gene NLRP3, enquanto os genótipos patogênicos são bem caraterizados, as variantes de baixa penetrância representam um desafio clínico, pois também podem ser encontradas em pacientes assintomáticos35. Outro detalhe é que aproximadamente 40% dos pacientes com síndromes CAPS clinicamente reconhecidas podem ser geneticamente negativos26.
TRATAMENTO
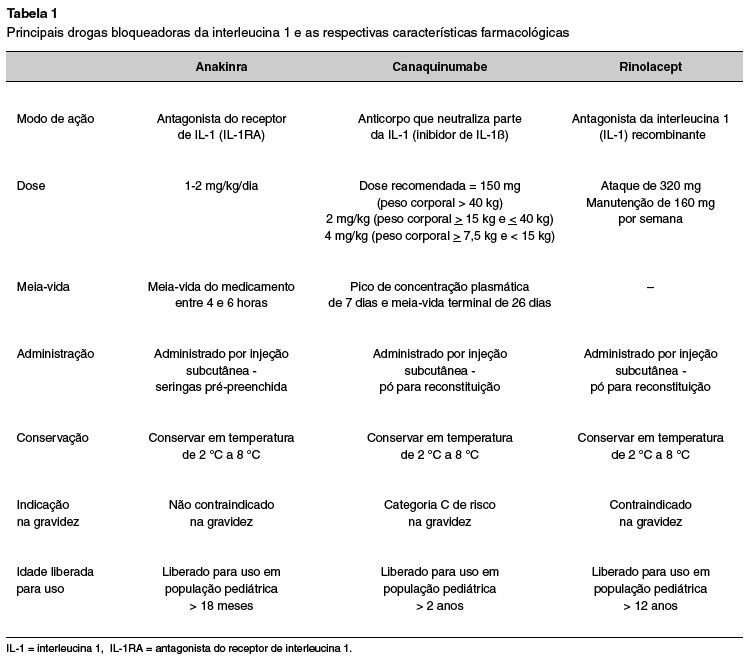
As terapias direcionadas à interleucina-1 consideradas efetivas no tratamento das CAPS-NLRP3 incluem: anakinra, rilonacept e o canaquinumabe1. O tratamento específico para inibição da atividade da interleucina-1 deve ser iniciado precocemente e, se possível, introduzido já em neonatos com sinais clínicos de síndrome CINCA grave, visando o controle rápido da inflamação e da incapacidade para o resto da vida27. A Tabela 1 resume os medicamentos disponíveis para tratamento das CAPS e suas principais características.
Anakinra
O anakinra é uma versão recombinante não glicosilada do antagonista do receptor de IL-1 (IL-1RA) preparado a partir de culturas de Escherichia coli geneticamente modificadas usando tecnologia de DNA recombinante. Sua estrutura é composta de 153 aminoácidos e difere do IL-1RA humano nativo por ter a adição de um único resíduo de metionina em seu amino terminal. O medicamento demonstrou controlar muitas doenças além das CAPS, e atualmente domina o campo da terapêutica com IL-1, principalmente em função de seu excelente perfil de segurança e tolerabilidade por pacientes pediátricos e adultos. Possui uma meia-vida entre 4 e 6 horas e requer aplicação diária pela via subcutânea. Há associação direta do mesmo com melhorias na qualidade de vida dos pacientes portadores de SMW e CINCA, propiciando alívio significativo dos sintomas clínicos sem que outras drogas imunossupressoras sejam necessárias. Embora o bloqueio de IL-1 não reverta os danos visuais e auditivos que tenham ocorrido antes do tratamento, o anakinra pode prevenir a progressão das lesões dos órgãos36,37.
O medicamento não possui alteração de efeito em gestantes, além de não ser comum a presença de prejuízo significativo para a saúde das mães ou dos fetos pela continuação do uso do anakinra durante a gestação. Em função da possibilidade de agenesia de órgãos e sua capacidade de passar através da placenta e entrar na mama, aumentos na dosagem de anakinra devem ser discutidos38.
A primeira experiência de uso foi relatada por Hawkins e cols., em 2003 e 2004. Cinco pacientes com MWS foram tratados com anakinra por vários meses e se observou um efeito rápido e substancial da droga em manifestações clínicas e biológicas39. Outros resultados animadores foram reportados posteriormente em todos os fenótipos de CAPS, ainda que estes relatos sejam baseados em estudos abertos e não randomizados40,41. Outros estudos iniciais de prova de conceito com o anakinra lograram melhorias importantes no tratamento de CAPS, evidenciando o papel crítico da IL-1 nestas doenças. Em curto prazo (52 meses), foi observada uma eficácia considerada alta durante estudo no qual 75% dos pacientes em tratamento com anakinra obtiveram remissão completa41.
Já com Chang e cols., o anakinra foi administrado durante a gravidez em 9 pacientes com CAPS (4 com FCAS, 1 com SMW e 4 com NOMID). Seis nascimentos a partir de mães FCAS e três nascimentos de mães NOMID ocorreram enquanto as pacientes estavam recebendo anakinra. Além disso, havia uma gravidez gemelar em que a mãe recebeu dose aumentada do medicamento. Nenhum parto prematuro ou complicações graves da gravidez foram observados. Um feto da gravidez de gêmeos teve agenesia renal e sofreu morte fetal40. Testes genéticos mostraram que o gêmeo falecido carregava a mesma mutação NLRP3 c.785T> C, p.V262A da mãe, e o outro gêmeo é saudável e não possui a mutação. O medicamento foi administrado de forma contínua durante as gestações e, além de ter sido bem tolerado, proporcionou alívio significativo e duradouro quanto aos sintomas de CAPS, assim como preveniu sequelas a longo prazo. Embora eficaz, o anakinra deve ser administrado diariamente e sua suspensão leva a um surto imediato da doença14.
Canaquinumabe
Canaquinumabe é um anticorpo que neutraliza parte da IL-1 e é considerado como uma droga útil para tratar qualquer fenótipo de CAPS. Este medicamento bloqueia seletivamente a interleucina-1β e não tem reatividade cruzada com outros membros característicos da família interleucina-1, incluindo interleucina-1α e interleucina-1Ra. Canaquinumabe é um anticorpo monoclonal humano, tem um período de semidesintegração longo de 23 a 26 dias e para atingir níveis estáveis no sangue leva cerca de oito semanas42,43. Em doses baixas a droga é utilizada para tratar as formas mais leves de CAPS, como FACS e MWS44, melhorando os sintomas como febre e urticária e reduzindo os marcadores inflamatórios. Contudo, esse medicamento não é suficiente para controlar a doença CINCA/NOMID mesmo com melhoria dos sintomas45. Comparado com outras moléculas monoclonais, o canaquinumabe tem uma ação de duração prolongada e uma baixa incidência de efeitos colaterais. Por outro lado, anakinra e rilonacept são frequentemente associados a efeitos colaterais requerendo um controle mais frequente, além de que o anakinra requer aplicação diária46.
Outras moléculas e terapia gênica
Rilonacept é uma proteína dimérica de fusão constituida por domínios de ligação de porções extracelulares de componentes do receptor para IL-1 (IL-R1 e proteína acessória do receptor da IL-1) acoplados à porção Fc da imunoglobulina humana IgG1 que se liga e neutraliza a IL-1. Foi a primeira droga autorizada para tratar as formas mais leves de CAPS nos Estados Unidos.
Hoje em dia, o rilonacept é usado com menos frequência devido aos efeitos secundários, tais como reações no local da injeção, sintomas respiratórios, cefaleia, náusea e hipertensão47. Uma outra droga é o MCC950, um inibidor de NLRP3 que reduz a produção de IL-1, mas ainda é pouco conhecida48.
A terapia gênica é uma das potenciais formas de tratamento das doenças monogênicas. Contudo, em formas genéticas de ganho de função há diversos fatores impeditivos, e ainda não há trabalhos em pacientes com doenças mediadas pelo gene NLRP3, NLRP12 ou NLRC4. De qualquer forma, os antagonistas da IL-1 melhoram a qualidade de vida dos pacientes sob o ponto de vista social e físico.
PROGNÓSTICO E ORIENTAÇÃO GENÉTICA
Amiloidose
Em FCAS, há um risco pequeno de desenvolvimento de complicações graves como a amiloidose, e neste grupo de pacientes a amiloidose renal, por exemplo, é vista infrequentemente, somente em torno de 2% dos pacientes14. Já a MSW apresenta maior número de crises persistentes e não relacionadas à exposição ao frio, o que acarreta um risco maior para desenvolvimento de amiloidose; esta doença acomete em média 25% dos pacientes com SMW, refletindo a intensa e prolongada resposta da fase aguda neste distúrbio49. Tem-se, assim, a amiloidose secundária como principal complicação da síndrome de Muckle-Wells. Na síndrome CINCA, esta mesma complicação pode chegar a 40% dos pacientes39.
Surdez
A perda de audição é associada a inflamações crônicas ou recorrentes que podem causar dano irreversível no ouvido interno com a perda de audição permanente2. Pacientes com MWS geralmente desenvolvem a perda auditiva neurossensorial com mais frequência, comparando com FACS17.
Orientação genética
Por se tratar de doença de herança autossômica dominante, todos os pacientes com mutações identificadas devem seguir com segregação familiar, análise genética dos familiares para procura da mesma mutação. Também, naqueles indivíduos cuja análise genética for negativa, vale ressaltar a pesquisa de mutações somáticas como causa da doença.
CONCLUSÃO
As síndromes periódicas associadas à criopirina (CAPS) são doenças de manifestações multissistêmicas decorrentes de mutações ao longo do gene NLRP3. A interleucina-1 desempenha papel fundamental na sua imunofisiopatologia e o bloqueio desta única citocina desempenha papel fundamental no tratamento destas doenças. Contudo, a resposta aos vários tipos de drogas capazes de bloquear a interleucina-1 varia de acordo com os diferentes fenótipos clínicos apresentados pelos pacientes. As principais complicações das CAPS são perda da qualidade de vida, surdez neurossensorial e amiloidose. Fenótipos mais graves, principalmente a síndrome CINCA, desenvolvem alterações de natureza osteoarticular devido ao supercrescimento ósseo e inflamações de cartilagens. Novas moléculas, bem como novos avanços na terapia gênica, poderão trazer maiores benefícios no tratamento das síndromes com natureza monogênica.
Referências
1. Yu JR, Leslie KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11(1):12-20. doi: 10.1007/s11882-010-0160-9.
2. Booshehri LM, Hoffman HM. CAPS and NLRP3. J Clin Immunol. 2019;39(3):277-286. doi: 10.1007/s10875-019-00638-z.
3. Xia X, Dai C, Zhu X, Liao Q, Luo X, Fu Y, et al. Identification of a Novel NLRP12 Non-sense Mutation (Trp408X) in the Extremely Rare Disease FCAS by Exome Sequencing. PLoS One. 2016;11(6):e0156981. doi: 10.1371/journal.pone.0156981.
4. Jéru I, Duquesnoy P, Fernandes-Alnemri T, Cochet E, Yu JW, Lackmy-Port-Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA. 2008;105(5):1614-9. doi: 10.1073/pnas.0708616105.
5. Jeskey J, Parida A, Graven K, Hostoffer R. Novel gene deletion in NLRC4 expanding the familial cold inflammatory syndrome phenotype. Allergy Rhinol (Providence). 2020;11:2152656720928062. doi: 10.1177/2152656720928062.
6. de Torre-Minguela C, Mesa Del Castillo P, Pelegrín P. The NLRP3 and Pyrin Inflammasomes: Implications in the pathophysiology of autoinflammatory diseases. Front Immunol. 2017;8:43. doi: 10.3389/fimmu.2017.00043.
7. Xiao J, Wang C, Yao JC, Alippe Y, Xu C, Kress D, et al. Gasdermin D mediates the pathogenesis of neonatal-onset multisystem inflammatory disease in mice. PLoS Biol. 2018;16(11):e3000047. doi: 10.1371/journal.pbio.3000047.
8. Carta S, Penco F, Lavieri R, Martini A, Dinarello CA, Gattorno M, et al. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc Natl Acad Sci USA. 2015;112(9):2835-40. doi: 10.1073/pnas.1424741112.
9. Lu A, Wu H. Structural mechanisms of inflammasome assembly. FEBS J. 2015;282(3):435-44. doi: 10.1111/febs.13133.
10. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16(7):407-20. doi: 10.1038/nri.2016.58.
11. Strowig T, Henao-Mejia J, Elinav E, Flavell R. Inflammasomes in health and disease. Nature. 2012;481(7381):278-86. doi: 10.1038/nature10759.
12. Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21(7):677-87. doi: 10.1038/nm.3893.
13. Campbell L, Raheem I, Malemud CJ, Askari AD. The Relationship between NALP3 and Auto-inflammatory Syndromes. Int J Mol Sci. 2016;17(5):725. doi: 10.3390/ijms17050725.
14. Goldbach-Mansky R, Shroff SD, Wilson M, Snyder C, Plehn S, Barham B, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58(8):2432-42. doi: 10.1002/art.23620.
15. Stych B, Dobrovolny D. Familial cold autoinflammatory syndrome (FCAS): characterization of symptomatology and impact on patients' lives. Curr Med Res Opin. 2008;24(6):1577-82. doi: 10.1185/03007990802081543.
16. Hedrich CM, Bruck N, Paul D, Hahn G, Gahr M, Rösen-Wolff A. "Mutation negative" familial cold autoinflammatory syndrome (FCAS) in an 8-year-old boy: clinical course and functional studies. Rheumatol Int. 2012;32(9):2629-36. doi: 10.1007/s00296-011-2019-3.
17. Kitley JL, Lachmann HJ, Pinto A, Ginsberg L. Neurologic manifestations of the cryopyrin-associated periodic syndrome. Neurology. 2010;74(16):1267-70. doi: 10.1212/WNL.0b013e3181d9ed69.
18. Biswas D, Stafford N. Otolaryngological manifestations of 'Muckle-Wells syndrome'. Int J Pediatr Otorhinolaryngol. 2010;74(5):553-5. doi: 10.1016/j.ijporl.2010.02.023.
19. Koitschev A, Gramlich K, Hansmann S, Benseler S, Plontke SK, Koitschev C, et al. Progressive familial hearing loss in Muckle-Wells syndrome. Acta Otolaryngol. 2012;132(7):756-62. doi: 10.3109/00016489.2012.656321.
20. Headley AP, Cordingley F, Hawkins PN, Riminton DS. Muckle-Wells cryopyrinopathy: complex phenotyping and response to therapy in a new multiplex kindred. Inflammation. 2014;37(2):396-401. doi: 10.1007/s10753-013-9752-6.
21. Thomas J, Maramattom BV. Muckle-Wells syndrome associated with multifocal clinically isolated syndrome. Int J Rheum Dis. 2014;17(3):349-50. doi: 10.1111/1756-185X.12317.
22. Tran TA, Koné-Paut I, Marie I, Ninet J, Cuisset L, Meinzer U. Muckle-Wells syndrome and male hypofertility: a case series. Semin Arthritis Rheum. 2012;42(3):327-31. doi: 10.1016/j.semarthrit.2012.03.005.
23. Finetti M, Omenetti A, Federici S, Caorsi R, Gattorno M. Chronic Infantile Neurological Cutaneous and Articular (CINCA) syndrome: a review. Orphanet J Rare Dis. 2016;11(1):167. doi: 10.1186/s13023-016-0542-8.
24. Sibley CH, Plass N, Snow J, Wiggs EA, Brewer CC, King KA, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012;64(7):2375-86. doi: 10.1002/art.34409.
25. Navallas M, Inarejos Clemente EJ, Iglesias E, Rebollo-Polo M, Zaki FM, Navarro OM. Autoinflammatory diseases in childhood, part 1: monogenic syndromes. Pediatr Radiol. 2020 Mar;50(3):415-30. doi: 10.1007/s00247-019-04536-9.
26. Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep. 2011;13(2):123-31. doi: 10.1007/s11926-011-0165-y.
27. Paccaud Y, Berthet G, Von Scheven-Gête A, Vaudaux B, Mivelaz Y, Hofer M, et al. Neonatal treatment of CINCA syndrome. Pediatr Rheumatol Online J. 2014;12:52. doi: 10.1186/1546-0096-12-52.
28. Almeida de Jesus A, Goldbach-Mansky R. Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin Immunol. 2013;147(3):155-74. doi: 10.1016/j.clim.2013.03.016.
29. Sarrabay G, Grandemange S, Touitou I. Diagnosis of cryopyrin-associated periodic syndrome: challenges, recommendations and emerging concepts. Expert Rev Clin Immunol. 2015;11(7):827-35. doi: 10.1586/1744666X.2015.1047765.
30. Johnstone RF, Dolen WK, Hoffman HM. A large kindred with familial cold autoinflammatory syndrome. Ann Allergy Asthma Immunol. 2003;90(2):233-7. doi: 10.1016/S1081-1206(10)62147-3.
31. Nakanishi H, Prakash P, Ito T, Kim HJ, Brewer CC, Harrow D, et al. Genetic hearing loss associated with autoinflammation. Front Neurol. 2020;11:141. doi: 10.3389/fneur.2020.00141.
32. Sag E, Bilginer Y, Ozen S. Autoinflammatory diseases with periodic fevers. Curr Rheumatol Rep. 2017;19(7):41. doi: 10.1007/s11926-017-0670-8.
33. Marzano AV, Tavecchio S, Venturini M, Sala R, Calzavara-Pinton P, Gattorno M. Urticarial vasculitis and urticarial autoinflammatory syndromes. G Ital Dermatol Venereol. 2015;150(1):41-50.
34. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet. 2001;29(3):301-5. doi: 10.1038/ng756.
35. Tanaka N, Izawa K, Saito MK, Sakuma M, Oshima K, Ohara O, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum. 2011;63(11):3625-32. doi: 10.1002/art.30512.
36. Bachove I, Chang C. Anakinra and related drugs targeting interleukin-1 in the treatment of cryopyrin-associated periodic syndromes. Open Access Rheumatol. 2014;6:15-25. doi: 10.2147/OARRR.S46017.
37. Koné-Paut I, Lachmann HJ, Kuemmerle-Deschner JB, Hachulla E, Leslie KS, Mouy R, et al. Sustained remission of symptoms and improved health-related quality of life in patients with cryopyrin-associated periodic syndrome treated with canakinumab: results of a double-blind placebo-controlled randomized withdrawal study. Arthritis Res Ther. 2011;13(6):R202. doi: 10.1186/ar3535.
38. Chang Z, Spong CY, Jesus AA, Davis MA, Plass N, Stone DL, et al. Anakinra use during pregnancy in patients with cryopyrin-associated periodic syndromes (CAPS). Arthritis Rheumatol. 2014;66(11):3227-32. doi: 10.1002/art.38811.
39. Hawkins PN, Bybee A, Aganna E, McDermott MF. Response to anakinra in a de novo case of neonatal-onset multisystem inflammatory disease. Arthritis Rheum. 2004;50(8):2708-9. doi: 10.1002/art.20357.
40. Rigante D, Leone A, Marrocco R, Laino ME, Stabile A. Long-term response after 6-year treatment with anakinra and onset of focal bone erosion in neonatal-onset multisystem inflammatory disease (NOMID/CINCA). Rheumatol Int. 2011;31(12):1661-4. doi: 10.1007/s00296-010-1787-5.
41. Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol. 2013;25(6):469-84. doi: 10.1016/j.smim.2013.10.008.
42. Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, Leslie KS, Hachulla E, Quartier P, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med. 2009;360(23):2416-25. doi: 10.1056/NEJMoa0810787.
43. Kuemmerle-Deschner JB, Ramos E, Blank N, Roesler J, Felix SD, Jung T, et al. Canakinumab (ACZ885, a fully human IgG1 anti-IL-1 mAb) induces sustained remission in pediatric patients with cryopyrin-associated periodic syndrome (CAPS). Arthritis Res Ther. 2011;13(1):R34. doi: 10.1186/ar3266.
44. Caorsi R, Lepore L, Zulian F, Alessio M, Stabile A, Insalaco A, et al. The schedule of administration of canakinumab in cryopyrin associated periodic syndrome is driven by the phenotype severity rather than the age. Arthritis Res Ther. 2013;15(1):R33. doi: 10.1186/ar4184.
45. Hoffman HM, Throne ML, Amar NJ, Cartwright RC, Kivitz AJ, Soo Y, et al. Long-term efficacy and safety profile of rilonacept in the treatment of cryopryin-associated periodic syndromes: results of a 72-week open-label extension study. Clin Ther. 2012;34(10):2091-103. doi: 10.1016/j.clinthera.2012.09.009.
46. Landmann EC, Walker UA. Pharmacological treatment options for cryopyrin-associated periodic syndromes. Expert Rev Clin Pharmacol. 2017;10(8):855-64. doi: 10.1080/17512433.2017.1338946.
47. Goldbach-Mansky R, Shroff SD, Wilson M, Snyder C, Plehn S, Barham B, et al. A pilot study to evaluate the safety and efficacy of the long-acting interleukin-1 inhibitor rilonacept (interleukin-1 Trap) in patients with familial cold autoinflammatory syndrome. Arthritis Rheum. 2008;58(8):2432-42. doi: 10.1002/art.23620.
48. Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med. 2015;21(3):248-55. doi: 10.1038/nm.3806.
49. van der Hilst JC, Simon A, Drenth JP. Hereditary periodic fever and reactive amyloidosis. Clin Exp Med. 2005;5(3):87-98. doi: 10.1007/s10238-005-0071-6.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888