Número Atual: Janeiro-Março 2021 - Volume 5 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
Atualizando e expandindo o universo de: "Uma nova classe de doenças - doenças autoinflamatórias"
Updating and expanding the universe of: "A new class of diseases - autoinflammatory disorders"
Leonardo Oliveira Mendonça1,2; Alex Isidório Prado1,2; Jorge Kalil1; Luiz Augusto Marcondes Fonseca1; Fábio Fernandes Morato Castro1; Myrthes Anna Maragna Toledo Barros1
1. Faculdade de Medicina da Universidade de São Paulo, Departamento de Imunologia Clínica e Alergia - São Paulo, SP, Brasil
2. Centro de Doenças Raras e da Imunidade, Hospital 9 de Julho - São Paulo, SP, Brasil
Endereço para correspondência:
Leonardo Oliveira Mendonça
Email: leonardo.oliveira.mendonca@gmail.com
Submetido em: 28/05/2020
Aceito em: 27/06/2020
RESUMO
As síndromes autoinflamatórias são doenças raras, genéticas de envolvimento prioritário da imunidade inata. Avanços nas técnicas de sequenciamento genético permitiram dissecar os genes envolvidos nestas doenças, continuamente organizando o quebra-cabeça genético e fisiopatológico de tais desordens. Este artigo revisa os últimos achados genéticos com seus respectivos fenótipos, código OMIM e ORPHA. Além disso, sugere cautela na triagem clínica e na indicação de métodos restritivos de sequenciamentos genéticos.
Descritores: Síndromes de imunodeficiência, doenças genéticas inatas, dados de sequência molecular, doenças hereditárias autoinflamatórias.
INTRODUÇÃO
Em julho de 2017, o artigo com título "Uma nova classe de doenças: doenças autoinflamatórias" foi publicado relatando um novo grupo de desordens imunológicas, enquadradas nos Erros Inatos da Imunidade, as doenças autoinflamatórias. Este grupo de doenças redefine as doenças inflamatórias sistêmicas, divididas, semanticamente, em dois grupos: autoimunes (relacionadas à imunidade adaptativa), e autoinflamatórias (relacionadas à imunidade inata).
A ferramenta que permitiu tal diferenciação, o sequenciamento genético, ganhou, nos últimos anos, grande disseminação e modernização, o que trouxe rapidez em realização e garante mais facilidade e confiabilidade nos resultados. Tal evolução facilitou não só o diagnóstico clinico e genético destas doenças, mas também, possibilitou e continua a garantir organização do quebra-cabeça genético e fisiopatológicos das doenças autoinflamatórias e diversas síndromes imunodesregulatórias.
Até então, as doenças autoinflamatórias eram classificadas em três grandes grupos, conforme a base genética e as consequentes ativações de vias intracelulares: as inflamossomopatias (doenças associadas ao inflamossoma); as proteossomopatias (doenças associadas ao protesossoma) e as relopatias (doenças associadas ao sistema NFκB).
No ano de 2020, o grupo italiano resumiu os 100 primeiros genes associados às doenças autoinflamatórias. Com base nesta revisão e dos recentes avanços do conhecimento dos processos imunológicos celulares decorrentes de mutações genéticas, uma nova classificação foi proposta.
Este artigo tem como função, além e atualizar tais achados, relatar a nova classificação das síndromes autoinflamatórias e as principais doenças dentro de cada grupo, com seus respectivos códigos OMIM e ORPHA.
MATERIAIS E MÉTODOS
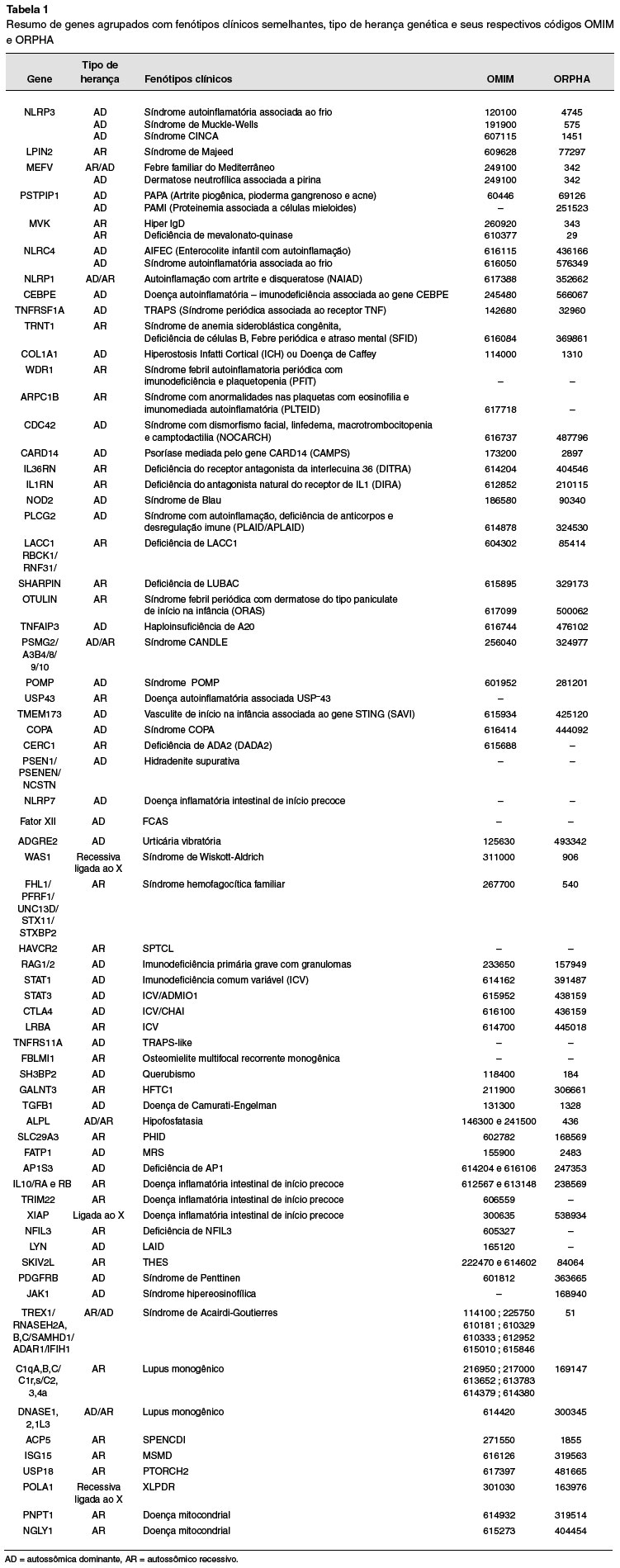
Foi utilizada a lista de genes envolvidas em síndromes autoinflamatórias conforme publicada por Pappa e cols., em 2020, como base para coleta de dados genéticos1. Foi realizada busca por todos os códigos OMIM e ORPHA conforme disponível on-line em https://www.omim.org/entry e https://www.orpha.net/consor, respectivamente. A lista de todos os genes foi agrupada em fenótipos semelhantes, quanto cabível. Os respectivos códigos, quando presentes, foram descritos de forma tabular.
RESULTADOS
O total de 100 genes foi reportado, contudo, quando agrupados em síndromes clínicas (fenótipos) semelhantes, 75 síndromes puderam ser classificadas. Do total, 10 genes não tinham fenótipo clínico associado na base de dados OMIM. Não respectivamente, outros 12 genes também não tinham fenótipos registrados na base de dados ORPHA. Somente 7 genes não tinham nem código OMIM e nem código ORPHA para correlação genotípica e fenotípica. Todos os genes, fenótipos e seus respectivos códigos estão resumidos na Tabela 1.
DISCUSSÃO
Com base nos resultados, um grande número de outras síndromes com fenótipos e genes anteriormente associadas a imunodeficiência primária ou a doenças autoimunes clássicas, apresentam, também, fenótipos autoinflamatórios, como por exemplo o gene WASP1 (síndrome de Wiskott-Aldrich) e C1qA (lupus eritematoso sistêmico). Estes achados ressaltam a necessidade de alerta para sinais precoces de manifestações inflamatórias como sinais iniciais de síndromes autoinflamatórias.
Outro fato é que o uso comercial de painéis genéticos restritos a poucos genes podem não englobar boa parte das síndromes já descritas, levando ao subdiagnóstico ou diagnósticos inconclusivos. Este fato ressalta a necessidade de extratificação clínica, laboratorial e imunológica cautelosa para guiar corretamente a solicitação e a interpretação de resultados genéticos.
As inflamossomopatias ocorrem da desregulação de complexos multiproteicos, chamados de inflamossomas, que culminam com secreção exagerada de interleucinas, IL1β e IL18. O acúmulo de proteínas mal formadas, que levam ao stress do retículo endoplasmático, como ocorre com o gene TNFRS1A, leva ao grupo das imuno-proteinopatias. Quando defeitos genéticos desregram a polimerização da actina, e consequentemente defeitos no citoesqueleto celular, reconhece-se as imunoactinopatias. Defeitos de sinalização celular, via sistema NFκB, como ocorre nas desordens clínicas associadas ao gene NOD2, ou através de defeitos na ubiquinização do proteossomo, são reconhecidas como relopatias. Por fim, um grupo de doenças cursa com defeitos na regulação do proteossoma, as interferonopatias.
Portanto, os últimos achados genéticos, além de permitir reorganizar tal grupo de síndromes, aprofundou os achados imunofisiopatológicos de diversas outras doenças, reagrupando, agora, em cinco grandes grupos, a saber: (1) Inflamossomopatias, (2) Proteossomopatias, (3) Imuno-proteinopatias, (4) Imuno-actinopatias, e (5) Relopatias (Figura 1).
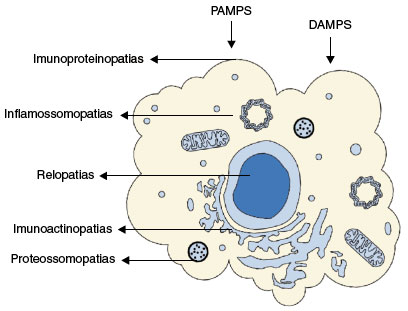
Figura 1
Figura ilustrativa do neutrófilos e componentes intracelulares. Os estímulos da imunidade inata através de DAMPs (Padrões Moleculares Associadas a Perigo) e PAMPs (Padrões Moleculares Associados a Patógenos) com as respectivas síndromes autoinflamatórias a depender do caminho imunológico hiperativado
Por último, vale ressaltar que aproximadamente 50% dos pacientes com síndromes clínicas compatíveis com doenças autoinflamatórias não possuem base genética identificada, o que não descarta uma doença autoinflamatória. Aproximadamente metade dos pacientes caracteriza-se ou por síndromes multifatoriais, ou por síndromes sem base genética.
CONCLUSÃO
As doenças autoinflamatórias, ou fenômenos autoinflamatórios, compõem número crescente de síndromes genéticas cujo diagnóstico clínico e genético ainda constitui grande desafio na prática clínica. Sugere-se ampla triagem clínica, laboratorial, imunológica e cautela na correlação genotípica e fenotípica, especialmente quando resultados duvidosos em painéis genéticos. O baixo número de relatos de casos na população brasileira reflete a necessidade de ampla divulgação e sinais de alerta para tais síndromes. Ressalta-se que parte considerável das síndromes inflamatórias sistêmicas não possui base genética.
AGRADECIMENTOS
A Douglas Amorim, pelo desenho da Figura 1.
REFERÊNCIAS
1. Papa R, Picco P, Gattorno M. The expanding pathways of autoinflammation: a lesson from the first 100 genes related to autoinflammatory manifestations. Adv Protein Chem Struct Biol. 2020;120:1-44. doi: 10.1016/bs.apcsb.2019.11.001.
2. https://www.omim.org/entry [site na Internet]. Acessado em 26/04/2021.
3. https://www.orpha.net/consor [site na Internet]. Acessado em 26/04/2021.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888