Número Atual: Julho-Setembro 2018 - Volume 2 - Número 3
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
- Outros do Autor

Artigo de Revisão
Corticoterapia
Corticotherapy
Hisbello S. Campos
DOI: 10.5935/2526-5393.20180050
Instituto Fernandes Figueira, Fiocruz, Serviço de Alergia e Imunologia Clínica - Rio de Janeiro, RJ
Endereço para correspondência:
p>Hisbello S. CamposE-mail: hisbello@globo.com
Submetido em: 22/06/2018
Aceito em 30/08/2018
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
Glicocorticosteroides são fármacos efetivos no tratamento de doenças inflamatórias e imunes. Agem em praticamente todas as células do corpo, antagonizando os efeitos patogênicos de inúmeras doenças. A maior parte de seus efeitos parece ser produto de sua ligação a receptores específicos armazenados no interior das células. Suas ações moduladoras da transcrição genética iniciam-se com a ligação ao seu receptor e posterior conexão aos genes alvo, num processo que conta com a participação de outros fatores e envolve múltiplos mecanismos (ação genômica). Os genes alvo incluem aqueles responsáveis por mediadores inflamatórios, como quimiocinas, citocinas, fatores de crescimento e seus receptores. Além de seus efeitos sobre o DNA, estimulando a produção de produtos anti-inflamatórios ou inibindo a transcrição de genes pró-inflamatórios, via acetilação ou deacetilação das histonas, respectivamente, os glicocorticosteroides possuem outros mecanismos de ação que não envolvem regulação genética (ação não genômica). Aparentemente, por mecanismos ainda não esclarecidos, os efeitos da corticoterapia são produto da associação das ações genômicas com as não genômicas. Os glicocorticosteroides representam o grande pilar terapêutico da asma, com efeitos sobre as células estruturais e funcionais do trato respiratório. Nessa situação particular, na qual costumam ser empregados continuadamente por períodos prolongados, com risco potencial de efeitos indesejáveis relevantes, é fundamental desvendar os processos envolvidos em seus mecanismos de ação para tentar desenvolver meios de reduzir os riscos associados e potencializar os efeitos desejados.
Descritores: Glicocorticosteroides, efeito genômico e não genômico, mecanismos de ação.
INTRODUÇÃO
O cortisol é um hormônio produzido na zona fasciculada das glândulas adrenais que controla inúmeras funções do nosso organismo, desde o metabolismo de carboidratos no fígado, até a imunidade e as respostas inflamatórias, que fazem parte das nossas defesas. Sua produção é regulada pelo eixo hipotálamo-hipófise-adrenal (HHA) e suas funções são regidas pelo receptor de glicocorticoide (RG)1.
Esse receptor está distribuído por todos os tecidos do corpo, havendo, no entanto, heterogeneidade na sua sensibilidade aos glicocorticosteroides (GC) e nas respostas biológicas induzidas por eles. A amplitude das ações dos GC sobre a biologia celular e seus efeitos anti-inflamatórios e imunossupressores fez deles instrumentos fundamentais no tratamento de diversas alterações inflamatórias agudas e crônicas, doenças autoimunes e hematológicas. Os GC sintéticos, usados como medicamentos (prednisolona, fluticasona, budesonida e dexametasona, p. ex.), são resultantes de modificações feitas na estrutura do cortisol (hidrocortisona) com o intuito de amplificar os efeitos anti-inflamatórios2 ou de reduzir os efeitos minerais, potencializar a deposição tópica e o metabolismo hepático3. Suas funções imunorregulatórias resultam tanto do efeito sobre o DNA, suprimindo a produção de proteínas pró-inflamatórias e estimulando a formação de proteínas anti-inflamatórias (efeito genômico), como de efeitos sobre outras estruturas celulares (efeito não-genômico). Caracteristicamente, o uso prolongado de GC é acompanhado por efeitos adversos, o que pode trazer inconvenientes graves em situações clínicas nas quais são úteis e, por vezes, fundamentais. Dessa forma, desvendar os mecanismos moleculares que regulam os efeitos fisiológicos dos GC pode se traduzir no emprego mais seguro desse grupo farmacológico. Pesquisas vêm sendo conduzidas visando desenvolver novos agonistas dos RG que conjuguem os mesmos efeitos imunossupressivos e anti-inflamatórios sem os efeitos prejudiciais ao organismo. No presente artigo, são apresentados alguns aspectos dos processos biológicos envolvidos na ação dos GC, com atenção particular em suas ações na asma.
CORTICOSTEROIDES
Os hormônios esteroides são gerados a partir do colesterol, e são divididos em mineralocorticoides, glicocorticosteroides (cortisol) e androgênios (17-cetosteroides), de acordo com a região do córtex da adrenal onde são produzidos. Os primeiros são produzidos na zona glomerulosa, e a aldosterona é o seu principal representante. Ela participa da regulação da homeostasia dos íons sódio e potássio, e, consequentemente, do volume intravascular. Sua secreção é primariamente regulada pelo sistema renina-angiotensina e pelas concentrações séricas de potássio. Quando ocorre redução do volume intravascular, da pressão de perfusão renal ou da carga filtrada de sódio, o aparelho justaglomerular renal produz renina, que transforma o angiotensinogênio produzido pelo fígado em angiotensina I, que é transformada em angiotensina II pela enzima conversora da angiotensina. A angiotensina II, ao se ligar a receptores específicos na zona glomerulosa da adrenal, estimula a produção da aldosterona. Os glicocorticosteroides (GC) são produzidos na zona fasciculada do córtex da adrenal, sendo a hidrocortisona (cortisol) o principal deles. Variações na molécula do cortisol dão origem aos demais GC, naturais ou sintéticos. Dentre os sintéticos, as modificações na molécula visam aumentar a potência anti-inflamatória e reduzir os efeitos indesejáveis, resultantes da atividade mineralocorticoide. A lipofilicidade que caracteriza os GC favorece sua absorção rápida após a administração tópica e permite que passem rapidamente pela membrana celular e entrem no citoplasma, onde se ligarão a receptores específicos que os levarão ao núcleo celular, onde exercerão seus efeitos junto ao DNA. Os GC inalatórios têm afinidade elevada pelo receptor, o que promove sua retenção nas vias aéreas. Por serem rapidamente metabolizados após sua absorção no trato gastrintestinal, têm perfil de segurança adequado4,5. Os hormônios androgênios (di-hidrotestosterona (DHT), testosterona, androstenediona e dehidroepiandrosterona (DHEA)) são sintetizados nas gônadas e nas adrenais a partir do colesterol. Apesar de a DHT ser mais potente que a testosterona, essa última é a que tem maior concentração sérica6. A testosterona e a androstenediona podem ser aromatizadas e convertidas em estrogênios pela aromatase7. Há evidências de que os androgênios afetem o desenvolvimento e ativação da célula imune, e há suspeitas de que diferenças na sua exposição entre os sexos estejam envolvidas na maior prevalência de doenças autoimunes em mulheres, e neoplasias em homens. A supressão da reatividade imune e inflamação induzidas pelos androgênios aumentaria os riscos para os dois grupos8.
Os GC são hormônios com ação em quase todas as células e tecidos do corpo. Podem ser administrados pelas vias oral (VO), intramuscular (IM), intravenosa (IV), intra-articular (IA) e tópica (cutânea, inalatória ou como colírio). Sua potência relativa depende da sua afinidade pelo receptor e de fatores intrínsecos. De acordo com a duração da sua ação, quando administrados pelas VO, IM, IV e IA, são classificados como de:
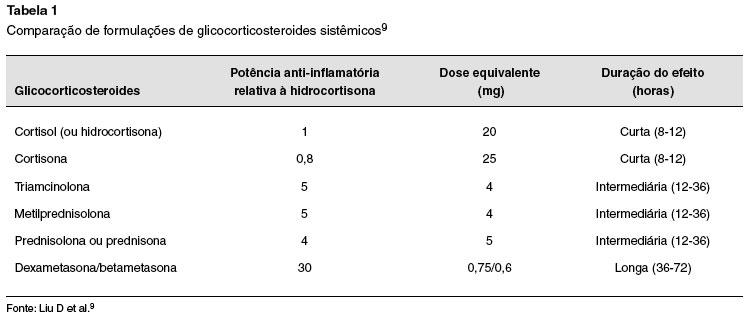
– ação curta: suprimem o ACTH por 8 a 12 horas. Cortisona e hidrocortisona;
– ação média: suprimem o ACTH por 12 a 36 horas. Prednisona, prednisolona, metilprednisolona e triancinolona;
– ação prolongada: suprimem o ACTH por 36 a 72 horas. Dexametasona e betametasona9.
Os GC atuam no metabolismo de carboidratos, lipídios e proteínas estimulando a gliconeogênese, aumentando a produção de glicogênio e também a sensibilidade dos tecidos à insulina. Ativam a lipólise e promovem a redistribuição de gordura. Também atuam sobre o sistema imune, inibindo a produção de citocinas que ativam o sistema imunitário, e diminuindo a expansão das células T e B. Seus efeitos anti-inflamatórios advêm do bloqueio ou retardo de etapas do processo inflamatório. Resumidamente, os efeitos dos GC são devidos à:
– redução dos linfócitos periféricos, particularmente as células T;
– inibição do acúmulo de neutrófilos no local da inflamação;
– apoptose das células linfoides;
– inibição da síntese de várias citocinas;
– modulação direta e indireta da função das células B;
– inibição da resposta proliferativa dos monócitos ao fator de estimulação de colônias;
– inibição da diferenciação de monócitos em macrófagos e da sua função fagocítica e citotóxica;
– inibição do movimento celular e dos fluidos a partir do compartimento intravascular;
– inibição da ação da histamina e dos ativadores do plasminogênio, e da síntese de prostaglandinas;
– ação bloqueadora da entrada da glicose nos tecidos;
– inibição da síntese proteica nos tecidos muscular, cutâneo, ósseo, conjuntivo, gorduroso e linfoide10.
Dependendo da molécula de GC, da dose e do tempo de uso, há risco variável de efeitos indesejáveis, que podem ser graves, fazendo com que seu uso por prazos longos deva ser cercado de cuidados e monitorado. Dentre eles, perda de massa muscular, conjuntiva e óssea, hipertensão arterial, hipercolesterolemia, fragilidade capilar, estrias cutâneas e catarata. Além disso, na infância, pode prejudicar o crescimento9.
PRODUÇÃO E REGULAÇÃO DOS GLICOCORTICOSTEROIDES
A regulação da produção de cortisol pela adrenal é feita pelo eixo hipotálamo-hipófise-adrenal (HHA). O cortisol passou a ser chamado glicocorticosteroide (GC) em função da sua capacidade de aumentar a disponibilidade de glicose no corpo humano. A produção dos esteroides pelas adrenais origina-se de uma reação em cascata no eixo HHA. O hipotálamo responde aos sinais derivados de fatores neurais, endocrínicos e de citocinas pró-inflamatórias11 que levam à ativação de neurônios contendo o Fator de liberação de corticotropina (CRF) nos núcleos paraventriculares. O CRF é carreado para a glândula pituitária anterior, onde estimula a produção e a liberação na corrente sanguínea do hormônio adrenocorticotrófico (ACTH ou corticotropina). O ACTH, por sua vez, estimula diretamente a córtex da adrenal para sintetizar e secretar GC (daí o termo corticosteroide) na circulação sistêmica. A ligação entre o eixo HHA e o relógio circadiano biológico confere o padrão diurno de secreção do GC, que faz com que sua concentração plasmática seja máxima de manhã, caia gradativamente durante o dia, e seja mais baixa à noite. A oscilação nos níveis plasmáticos é importante para os processos biológicos associados aos níveis de atividade do cotidiano. O eixo HHA também funciona como um componente do sistema de estresse sistêmico, o que provoca liberação de GC em situações de estresse físico ou emocional. Como referido anteriormente, citocinas pró-inflamatórias (IL-1, TNFα, IL-6 e interferons tipo I) também estimulam o hipotálamo a dar início ao processo de produção e liberação de GC. A liberação dessas citocinas como resposta a patógenos e/ou injúria tecidual atenua a resposta inflamatória e imune, completando um ciclo com feedback regulatório12.
A atividade e a disponibilidade do GC é regulada após sua secreção nos níveis teciduais e celulares. Em condições normais, 80-90% do GC circulante está ligado à globulina de ligação ao corticosteroide (Corticosteroid-binding globulin - CBG) e o restante à albumina. Dessa forma, a biodisponibilidade do GC depende da concentração de CBG, que age como um tampão contra picos de cortisol. No nível celular, a atividade do GC é regulada por enzimas da família das 11β-hidroxiesteroide desidrogenases (11β-HSD). Duas dessas enzimas têm papel antagônico, característica importante no processo de regulação. Enquanto a 11β-HSD1 facilita a conversão da cortisona em cortisol, amplificando a ação local do GC, a 11β-HSD2 faz o oposto, reduzindo a ação do hormônio13.
MECANISMO DE AÇÃO DOS GLICOCORTICOSTEROIDES
Há três mecanismos de ação conhecidos dos GC. Um deles, talvez o principal, é chamado genômico, por envolver efeitos sobre o DNA da célula alvo. Os outros dois são chamados não-genômicos, por não atuarem sobre o DNA. No mecanismo genômico, a ligação do GC ao seu receptor citoplasmático – receptor de glicocorticoide (RG) – dá início à maior parte dos efeitos celulares dos GC. O RG é um ligante regulado pelo fator de transcrição da superfamília dos receptores nucleares, que ativam ou reprimem a expressão de milhares de genes. O RG consiste numa estrutura proteica multifuncional que transcodifica o sinal emitido pelo GC; sua função é regulada pelo cortisol endógeno ou por GCs exógenos14. Até recentemente, acreditava-se que o RG estaria inativo no citoplasma celular, conjugado a um complexo proteico (chaperonas) constituído por proteínas de choque térmico (heat shock protein - hsp) e imunofilinas (FKBP51, FKBP52, Cyp44 e PP5)15, que conferem ao RG a forma tridimensional necessária para sua ligação ao GC. Atualmente, sabe-se que o RG tem fluxo contínuo entre os dois compartimentos celulares (citoplasma e núcleo), e que as proteínas chaperonas não estão restritas ao citosol16. O RG, como qualquer receptor nuclear, possui três domínios: (1) domínio de ligação ao ligante hormonal, localizado na extremidade carboxi-terminal; (2) domínio central de ligação ao DNA, que compreende o sítio de dimerização; e (3) domínio de transativação na extremidade aminoterminal, responsável pela interação com os fatores de transcrição7. O primeiro domínio tem como função ligar-se ao GC e promover a translocação nuclear do complexo GC-RG, a dimerização, a transativação do gene alvo e o silenciamento do receptor na ausência de ligação hormonal17. O segundo domínio é responsável pelo reconhecimento das sequências de ligação do RG nos genes alvos (elementos de resposta aos receptores de GC-RE). O terceiro domínio é responsável pela ligação do RG aos fatores de transcrição ou às moléculas coativadoras.
Como descrito anteriormente, em sua forma citoplasmática, o RG está ligado a um complexo multiproteico com chaperonas (hsp90 e hsp70). Ao se ligar ao GC, passa por alteração conformacional, expondo sinais de localização nuclear e, subsequentemente, desloca-se para o núcleo da célula18. Nesse processo de dissociação do RG do complexo proteico, o dímero CG-RG migra para o núcleo da célula, onde gerará os efeitos genômicos; o complexo proteico passará a integrar processos de sinalização que darão origem aos efeitos rápidos não-genômicos. No núcleo da célula, o complexo GC-RG dirige-se para a cromatina (complexo de DNA, RNA e proteínas, das quais as histonas são as principais). As histonas têm papel fundamental na estrutura e função das cromatinas, representando a espinha dorsal da cromatina e regulando a expressão dos genes, sua transcrição e supressão (silenciamento). A acetilação da histona é uma etapa importante no processo de transcrição. No processo de acetilação, as enzimas histona acetil transferase (HAT) dissociam o DNA do bloco proteico, desenrolando-o, expondo as áreas de ligação (GRE) ao complexo RG-GC e permitindo a transcrição. De modo inverso, o processo conjunto de metilação do DNA e a deacetilação da histona, através da ação das histonas deacetilases (HDACs), levam ao silenciamento do gene através de sinais que compactam o DNA em torno da histona, ocultando as áreas de ligação. Estima-se que os GC regulem de dez a cem genes por célula, diretamente, e muitos outros de modo indireto, através da interação com fatores de transcrição e coativadores19.
EFEITOS GENÔMICOS DOS GLICOCORTICOSTEROIDES
Acredita-se que, no núcleo, o complexo GC-RG se ligue e interaja com áreas específicas do DNA, denominadas elemento de resposta aos GC (GRE), via múltiplos mecanismos dependentes e independentes da ligação ao DNA, gerando resposta genômica20. Essas áreas de ligação direta do RG ao DNA são classificadas como “positivas (GRE+)” ou “negativas (GRE- ou GREn)”, dependendo da expressão dos genes alvos. A ligação numa área GRE+ do gene alvo estimularia a expressão do gen; se num local GREn, reprimiria a transcrição do gene alvo. Essa visão tradicional está sujeita a discussão, dado que sabemos que genes não são simplesmente “ligados” ou “desligados”. Na verdade, por mecanismos ainda não esclarecidos, a expressão de um gene é finamente ajustada para atingir as necessidades da célula. Ao “escolher” o local no DNA onde se ligará e dará início a uma série de atividades transcricionais ou supressivas, o RG “define” os genes alvos e como eles serão modulados21. A própria ligação do complexo GC-RG-GRE à uma área específica de um gene no DNA é um processo complexo que, certamente, envolve mecanismos ainda não esclarecidos que devem contar com a participação de outros fatores, como ligantes e coativadores tanto na ativação22 como na repressão23 genética. Os GRE consistem de duas metades de cadeias com seis nucleotídeos fixos separadas por três bases variáveis, nas quais o GC se liga diretamente como um homodímero24. As variações nos GRE levam a diferentes requerimentos de cofatores, diferentes níveis de transativação gênica e programas transcricionais diversos25.
O RG ativado tem a capacidade de associar-se a outro fator de transcrição ligado ao mesmo GRE e regular sua atividade transcricional. Esse mecanismo de regulação gênica pode se dar de duas maneiras. Numa, não é necessária a interação direta do RG com o DNA; a transrepressão envolve ligação direta proteína-proteína do RG a outros fatores de transcrição e interferência com seus mecanismos de ação26. Em outra, o RG liga-se ao DNA através do GRE e, por meio de seu terceiro domínio, também se conecta a um fator de transcrição adjacente e ligado ao DNA, afetando sua atividade transcricional de modo composto. Essa segunda maneira é a empregada na repressão da transcrição de fatores pró-inflamatórios, como o Fator nuclear kappa-κ (NF-κB) e a proteína ativadora-1 (AP-1)27, bem como na modulação da atividade de certos membros da família de transdutores de sinal e ativadores da transcrição (STAT)28. Esses fatores pró-inflamatórios estão ligados a sequências específicas de reconhecimento na região promotora dos genes inflamatórios, e agem como teclas moleculares que controlam a transcrição genética e têm atividade sobre a HAT, acetilando as histonas e reduzindo sua carga, o que permite que a estrutura da cromatina passe da conformação de repouso, fechada, para a forma aberta, ativada29. Isso resulta no desenrolar do DNA do bloco proteico (histonas), e subsequente ligação a fatores associados e à RNA polimerase II, iniciando a transcrição do gene. A ativação dos genes pelos GC está associada à acetilação seletiva de resíduos de lisina na histona, o que resulta em aumento da transcrição genética30. Esse mecanismo molecular é comum a todos os genes, incluindo os envolvidos na diferenciação, proliferação e ativação celular. Esse processo é reversível, e a deacetilação das histonas está associada ao silenciamento do gene. Além disso, é mediado pelas HDACs, que agem como corepressoras, em conjunto com outras proteínas corepressoras recrutadas a seguir. Resumidamente, os genes inflamatórios e os anti-inflamatórios são regulados pela acetilação e deacetilação/metilação das histonas, respectivamente.
O papel das histonas é amplo, funcionando, também, como reguladores essenciais nos processos epigenéticos ao mediar mudanças dinâmicas na diferenciação e aquisição de funções efetoras de células T CD4+. Esse reconhecimento faz das HDAC alvos terapêuticos potenciais nas doenças imunes mediadas por células T31. A evolução do conhecimento na área da genética vem apontando para novas perspectivas terapêuticas nas doenças autoimunes e em outras nas quais o comportamento celular alterado compõe a base patogenética, como a asma e o câncer, por exemplo. Nessa linha, fármacos epigenéticos que atuem sobre enzimas responsáveis pela transcrição ou repressão (DNA metiltransferase, demetilases Tet, lisina/arginina metiltransferases, lisina demetilase, HAT e HDAT) que atuam nos nucleossomas vêm sendo desenvolvidos e testados. Através da inibição dessas enzimas, pode-se atuar sobre cofatores importantes na determinação de comportamentos celulares indesejáveis32. A acetilação da lisina é uma modificação reversível pós-tradução das proteínas celulares que representa uma mudança regulatória importante nas cascatas de transdução de sinais33. A ativação da lisina, em combinação com outras modificações pós-tradução, direciona os desfechos e os níveis de ativação de mecanismos de transdução de sinais, como os do NFκB, por exemplo. Inibidores das HATs (transcritoras), e das HDACs (repressoras), têm o potencial de regular o NFκB, intervindo terapeuticamente nas doenças inflamatórias34.
O processo de ligação do RG a sequências específicas dos genes alvos e consequente modulação positiva ou negativa da expressão de genes também depende de fatores estruturais associados ao domínio de ligação do RG, onde existe um local específico no qual o GC se liga35. A afinidade, duração de ação e perfil de efeitos indesejáveis depende do grau de ocupação desse local. Essa propriedade é empregada no desenho das estruturas químicas dos GC sintéticos, visando amplificar os efeitos desejáveis e a segurança4.
EFEITOS NÃO-GENÔMICOS DOS GLICOCORTICOSTEROIDES
Como nem todas as ações dos GC podem ser explicadas por seus efeitos sobre o DNA, postulou-se que uma parcela das ações seria resultado de outros mecanismos, que não envolveriam o genoma. A partir daí, passou-se a classificar como efeitos não-genômicos qualquer ação que não afete a expressão gênica direta ou indiretamente, mas que induza efeitos rápidos, como a ativação de processos envolvidos com a ativação de sinais. O fato de serem rápidos implica que os efeitos não sejam afetados por inibidores de transcrição e síntese proteica. Ficou definido que esses efeitos de sinalização rápida acontecem na membrana celular. Os dois mecanismos principais de ações não-genômicas dos GC estariam associados à membrana plasmática, e aos canais iônicos. O primeiro deles envolveria o RG clássico, porém ligado à membrana plasmática, e não aquele situado no citoplasma, além de um segundo RG, diferente do clássico, e também localizado na membrana plasmática. O segundo mecanismo estaria ligado à entrada do cálcio no interior da célula36.
Na medida em que estudos específicos vêm trazendo mais luz sobre o tema, passamos a compreender que a sinalização genômica e não-genômica estão ligadas, e que as mudanças transcricionais em resposta aos GC requerem o funcionamento de ambos os processos. Atualmente, sabe-se que a sinalização não-genômica tanto pode ser iniciada no RG da membrana plasmática, como em outras moléculas de sinalização da mesma membrana. Os sinais não-genômicos podem modular a transcrição nuclear por diferentes mecanismos, incluindo alterações na fosforilação de moléculas importantes de sinalização nuclear37. Como exemplo da associação de efeitos genômicos e não-genômicos, pode-se citar os mecanismos envolvidos na ação da aldosterona. Suas ações regulatórias da pressão sanguínea e da homeostase hidroeletrolítica são independentes de outros efeitos sobre os tecidos cardiovasculares, como inflamação, fibrose, hipertrofia e remodelamento. Além dos efeitos genômicos, a aldosterona produz efeitos rápidos, em minutos, que não requerem transcrição ou tradução gênica, mediados por receptores de mineralocorticoides (RM) clássicos, e envolvendo diferentes processos de sinalização. Localizados próximos à membrana plasmática, os RM parecem estar associados a outros receptores (receptores de tirosinoquinases – EGFR, PDGFR, 1GF1R – e receptores acoplados à proteína G – AT1 e GPER1) que mediam os efeitos não-genômicos da aldosterona. Aparentemente, há interação e sinergismo entre as sinalizações genômica e não-genômica38. Dessa forma, desvendar os mecanismos envolvidos na sobreposição das ações genômicas e não-genômicas dos GC é uma etapa fundamental no desenvolvimento de fármacos que preservem/amplifiquem as ações desejadas e inibam/bloqueiem as indesejadas dos GC.
EMPREGO CLÍNICO DOS GLICOCORTICOSTEROIDES
A via de administração do GC depende da doença a ser tratada. Eles são efetivos pelas vias oral, intramuscular, intravenosa, inalatória e tópica. A dose farmacológica é aquela necessária para manejar a condição que requer a supressão da inflamação ou da alteração imune, e depende da farmacocinética das diferentes preparações de GC, dos efeitos das alterações clínicas subjacentes, e das interações com outros fármacos que poderão ser usados em paralelo. As diferentes formas de preparação dos GCS são bioequivalentes entre si, isso é, as formulações orais têm taxas de absorção e biodisponibilidade sistêmica semelhantes. O mesmo vale para as formulações tópicas ou injetáveis. No caso dos GC inalatórios, a biodisponibilidade varia de acordo com as propriedades físicas de cada fármaco. Classicamente, as diferentes formulações de GC empregados pela via sistêmica são comparadas tomando por base suas potências anti-inflamatórias relativas às da hidrocortisona e as equivalências entre suas doses (Tabela 1)9.
EFEITOS DOS GLICOCORTICOSTEROIDES NA IMUNIDADE
Reconhecidamente, ao mesmo tempo que antagonizam inflamação, os GC interferem com a função imune. A influência do sistema imune sobre o eixo HHA foi identificada quando foi detectado que infecções estavam associadas à ativação do eixo. Nessas situações, aparentemente, o agente imune mais potente é a IL-139. Outras citocinas, como a IL-6, IL-10 e o fator de necrose tumoral-alfa (TNF-α) também parecem ser dotados dessa propriedade, mas de forma menos potente que a IL-140. Entretanto, os mecanismos envolvidos no efeito supressor da imunidade dos GC ainda não estão totalmente esclarecidos. Há indícios de que, inicialmente, a ligação do GC ao RG afete as funções de proteínas intracelulares importantes para a função das células imunes. Por exemplo, o GC suprime a função do NF-κB através da indução da proteína inibitória 1κBα41. Como a AP-1 também parece implicada no efeito imunossupressor dos GC, possivelmente há diferentes mecanismos envolvidos nesse efeito dos GC42. Outras linhas de investigação indicam que o GC, associado à vitamina D43 ou ao ligante CD4044, geraria células dendríticas tolerogênicas que requerem expressão elevada de IL-10 ou PDL-1 para induzir Tregs, com consequente inibição de células T CD4+ ativadas. Resumidamente, essas linhas de estudo sugerem que os efeitos dos GC induziriam um fenótipo anti-inflamatório estável sobre os monócitos, suprimindo a proliferação de células T e a liberação de citocinas pelas células T CD8+ e T CD4+, particularmente essas últimas. Assim, os monócitos ativados pelos GC usariam dois mecanismos distintos para controlar e regular as células T CD4+. Num, rápido e eficiente, haveria a inibição direta das células T ativadas, sem a participação dos Treg. No outro, induziriam Tregs localmente nas áreas inflamadas45.
GLICOCORTICOSTEROIDES NA ASMA
Na asma, o uso regular de GC tem efeitos antiinflamatórios sobre, praticamente, todas as células do trato respiratório implicadas na inflamação asmática. Os GC têm efeito sobre as células imunes que infiltram o pulmão asmático e sobre a função das células estruturais das vias aéreas. Previnem o recrutamento de eosinófilos a partir da medula óssea para as vias aéreas, e suprimem a expressão de fatores de sobrevivência dessas células, ao mesmo tempo em que induzem sua apoptose46,47. Reduzem a expressão de citocinas pró-inflamatórias e quimiocinas dos macrófagos48 e a concentração sérica de monócitos, além reduzir a expressão dos receptores de IgE49. Eles inibem a ativação linfocitária e a expressão de mediadores inflamatórios através de diversos mecanismos, além de induzirem a apoptose do linfócito50. Seu emprego regular reduz marcadores inflamatórios observados nos brônquios, melhora a função pulmonar e, naqueles indivíduos com formas leves a moderadas da asma, reduz a hiper-responsividade brônquica (HRB). Entretanto, os efeitos benéficos dos GC no trato respiratório não atingem todos os asmáticos nem são permanentes, e não têm o potencial de curar a asma51.
Fatores genéticos estão envolvidos tanto na determinação da asma e das suas diferentes apresentações, como na resposta aos corticosteroides inalatórios (CSi). Numerosos estudos identificaram diversos polimorfismos genéticos que podem influenciar a resposta clínica aos CSi em crianças asmáticas. Os efeitos diretos ou indiretos dependem do papel dos polimorfismos no processo inflamatório asmático, ou sobre os mecanismos de ação antiinflamatórios dos GC. No primeiro grupo, os genes receptor 1 do hormônio liberador de corticotropina (CRHR1), membro 1 do grupo C da subfamília 3 do receptor nuclear (NR3C1), fosfoproteína 1 induzida por estresse (STIP1), fosfatase 1 de especificidade dupla (DUSP1), induzido por glicorticoide 1 (GLCCI1), histona deacetilase 1 (HDAC1), regulador 3 da biossíntese de esfingolipídio ORMDL (ORMDL3) e fator de crescimento epitelial vascular (VEGF), afetam diretamente a variabilidade da resposta anti-inflamatória aos CSi. No segundo grupo, dos efeitos indiretos, variantes no T-box 21 (TBX21) e o fragmento Fc do receptor II de IgE (FCER2) contribuem indiretamente para a variabilidade na resposta aos CSi, alterando os mecanismos inflamatórios da asma52. As consequências práticas da identificação desses polimorfismos seriam as predições das respostas aos CSi em crianças asmáticas, aumentando o valor potencial da abordagem terapêutica selecionada.
Fatores genéticos também estão envolvidos na resposta aos CSi quando usados em associação aos beta-2 agonistas de longa duração (LABA). Quando estudos clínicos indicaram que associar um LABA é mais efetivo que aumentar a dose do CSi53, começaram outros estudos procurando identificar os mecanismos subjacentes desse benefício. Aparentemente, o sinergismo entre ambos tem origem em múltiplas ações genéticas, ainda não totalmente esclarecidas, que incluem a promoção de genes anti-inflamatórios induzida tanto pelo GC como pelo LABA. Outros genes, que só são estimulados pelo uso conjunto dos dois medicamentos, também participam do efeito sinérgico. Dentre eles, ressalta o valor do fosfatase 1 especificidade dupla (DUSP1) na musculatura lisa peribrônquica e células epiteliais, e do sinalizador 2 regulador da proteína G (RGS2) no músculo liso49.
Alguns pontos referentes à corticoterapia inalatória na asma merecem comentários específicos. Um deles refere-se aos efeitos adversos, posto que o GC é usado regularmente e por períodos prolongados (anos). A probabilidade de efeitos adversos sistêmicos com o uso regular de CSi é reduzida, mas a de efeitos locais é maior. Esses últimos, de menor gravidade e devidos à deposição do CSi na orofaringe e laringe, incluem disfonia (associada à miopatia dos músculos laríngeos, irritação da mucosa e candidíase laríngea)54 e candidíase de orofaringe55. Por outro lado, o risco de efeitos adversos sistêmicos com a corticoterapia inalatória regular é concreto, embora a discussão sobre eles deva sempre incluir sua relevância clínica. Esse risco é influenciado por diversos fatores, como a dose administrada, o local da deposição (boca, pulmão, trato gastrintestinal), o equipamento inalatório, fatores pessoais (idade, sexo, tabagismo, cálcio e vitamina D na dieta, atividade física), diferenças individuais na resposta aos GC e comorbidades56. A gravidade da asma é um outro fator modulador do risco de efeitos adversos sistêmicos. Entre os asmáticos graves, ele é influenciado de modo oposto por dois fatores. Por um lado, a maior limitação ao fluxo aéreo reduz sua deposição nas vias aéreas mais periféricas e, consequentemente, a absorção sistêmica. Por outro, a dose utilizada é maior, favorecendo o aumento da dose absorvida. Quando os efeitos sistêmicos dos CSi são colocados em pauta, a população de crianças e adolescentes asmáticos passa a ter interesse particular. Está bem estabelecido que seu uso é seguro nas doses recomendadas, mas, embora raramente, insuficiência adrenal aguda foi relatada em crianças usando doses acima das consideradas elevadas por prazos superiores a 6 meses e uso concomitante de GC nasal57.
Um outro ponto diz respeito ao risco potencial de infecções respiratórias em asmáticos que usam corticosteroide inalatório regularmente por prazos longos. Metanálise recente, analisando a associação de uso regular de CSi e risco de pneumonia e outras infecções respiratórias em crianças asmáticas, avaliou 39 ensaios, dos quais 31 incluíam 11.615 pacientes, comparando CSi por, pelo menos 4 semanas, com placebo. Mesmo ressaltando a falta de critérios claros para definir infecção respiratória, concluíram que o uso regular de CSi não aumentava o risco de pneumonia ou outras infecções respiratórias (faringite, otite e sinusite) em crianças com asma58.
O tamanho da partícula inalada é outro tópico que merece discussão. Esse debate surgiu com a demonstração de que a disfunção das pequenas vias aéreas periféricas (com diâmetro interno igual ou menor que 2 µm) está associada aos sintomas respiratórios e às características clínicas da asma59. Dessa forma, as pequenas vias aéreas tornaram-se um alvo importante do tratamento da asma, embora ainda haja debate na literatura sobre o valor clínico do tamanho das partículas geradas pelos inaladores atuais de uso médico. Basicamente, o debate principal foca as partículas finas e as extrafinas. Nessa área, o termo tamanho se refere à extensão linear da partícula, e o diâmetro da partícula é expresso como MMAD (diâmetro médio aerodinâmico da massa da gotícula). A partícula é chamada de fina quando o MMAD é < 5 µm. Quando o MMAD está em torno de 1 µm, a partícula é chamada de extrafina. Certamente, partículas menores têm a capacidade de penetrar mais profundamente as vias aéreas, exercendo seus efeitos sobre praticamente todas as ordens de vias aéreas intrapulmonares. Porém, inicialmente, sugeriu-se que partículas menores que 1 µm não seriam efetivas, já que seriam exaladas pelo paciente logo após inaladas60. Entretanto, alguns estudos contradizem essa suposição61, e ainda há debate sobre qual o tamanho ideal na faixa de 1 a 5 µm, ou seja, partículas finas ou extrafinas. Recentemente, busca sistemática na literatura e metanálise, avaliando estudos clínicos realizados entre 1998 e 2014, identificou 23 ensaios comparando os efeitos da inalação de propionato de fluticasona em partículas padrão, com aqueles obtidos com a inalação de partículas finas em crianças e adultos asmáticos. Os desfechos avaliados foram: volume expiratório forçado no primeiro segundo (VEF1), pico de fluxo expiratório (PFE) matinal, escore de sintomas, percentual do fluxo expiratório forçado predito entre 25 e 75% da capacidade vital (FEF25-75), e uso de medicação de resgate. O estudo concluiu que não havia diferença clinicamente significante na eficácia do tratamento entre os dois grupos62. No entanto, diversos outros estudos bem conduzidos apontaram que o emprego de partículas extrafinas traria desfechos melhores no tratamento de asmáticos55. Por exemplo, dois estudos demonstraram que sistemas inalatórios que geram partículas extrafinas de GC, com MMAD de aproximadamente 1 µm, depositam 50 a 60% da dose liberada, em contraste com sistemas que geram partículas com 3-4 µm, que depositam nos pulmões 10 a 20% da dose inalada63,64. Se considerado que os desfechos utilizados nos estudos sobre efetividade terapêutica em asmáticos são modulados por diversos fatores externos (técnicas e empenho pessoal na medida de parâmetros funcionais, percepção de sintomas, variações pessoais do comprometimento causado pela asma ao longo do tempo, acurácia das informações prestadas pelos pacientes, entre outras), o debate sobre qual o tamanho mais adequado da partícula medicamentosa inalada, assim como outros comparando efetividade terapêutica de diferentes medicamentos, estarão sempre longe da unanimidade.
Ainda considerando a via inalatória, os compostos com maior propriedade lipofílica (Fluticasona e Beclometasona) ficam retidos por períodos maiores no tecido pulmonar. Não se deve esquecer que uma parcela relevante da dose liberada pelo sistema de inalação é deglutida, e que uma parte da dose depositada no pulmão penetra a circulação sistêmica. Assim, 20-40% da dose administrada é absorvida65. Os GC são metabolizados no fígado e em outros tecidos pelo citocromo P450 3A4 (CYP 3A4), e por outras transformações (transportadores transmembrânicos da glicoproteína P)66. Dessa forma, medicamentos inibitórios ou indutores da CYP3A4 podem alterar a concentração sérica dos GC, incluindo os originalmente administrados pela via inalatória.
O FUTURO DOS GLICOCORTICOSTEROIDES
A compreensão crescente sobre a associação dos efeitos genômicos e não-genômicos nos efeitos anti-inflamatórios, imunossupressivos e antialérgicos vem orientando estudos visando desenvolver novos GC sintéticos cujos efeitos seriam resultantes, principalmente, de mecanismos não-genômicos. Dessa forma, o potencial de efeitos indesejáveis desses novos fármacos seria menor67. Outras classes terapêuticas, como fármacos epigenéticos, provavelmente agregarão vantagens relevantes no tratamento das doenças inflamatórias, reduzindo a grande dependência terapêutica nos glicocorticosteroides.
REFERENCES
1. Li D, Sánchez ER. Glucocorticoid receptor and heat shock factor 1: novel mechanism of reciprocal regulation. Vitam Horm. 2005;71:239-62.
2. Daley-Yates PT. Inhaled corticosteroids: potency, dose equivalence and therapeutic index. Br J Clin Pharmacol. 2015;80(3):372-80.
3. Hochhaus G, Druzgala P, Hochhaus R, Huang MJ, Bodor N. Glucocorticoid activity and structure activity relationships in a series of some novel 17 alpha-ether-substituted steroids: influence of 17 alpha-substituents. Drug Des Discov. 1991;8(2)117-25.
4. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711.
5. Hansel TT, Benezet O, Kafé H, Ponitz HH, Cheung D, Engelstätter R, et al. A multinational, 12-week, randomized study comparing the efficacy and tolerability of ciclesonide and budesonide in patients with asthma. Clin Ther. 2006;28(6):906-20.
6. Marchetti PM, Barth JH. Clinical biochemistry of dihydrotestosterone. Ann Clin Biochem. 2013;50:95-107.
7. Griffin JE, Wilson JD. The syndromes of androgen resistance. N Engl J Med. 1980;302:198-209.
8. Gubbels Bupp MR, Jorgensen TN. Androgen-Induced Immunosuppression. Front Immunol. 2018;9:794.
9. Liu D, Ahmet A, Ward L, Krishnamoorthy P, Mandelcorn ED, Leigh R, et al. A practical guide to the monitoring and management of the complications of systemic corticosteroid therapy. Allergy Asthma Clin Immunol. 2013,9:30.
10. Boardman C, Chachi L, Gavrila A, Keenan CR, Perry MM, Xia YC, et al. Mechanisms of glucocorticoid action and insensitivity in airways disease. Pulm Pharmacol Ther. 2014;29(2):129-43.
11. Webster JL, Tonelli L, Sternberg EM. Neurendocrine regulation of immunity. Annu Rev Immunol. 2002;20:125-63.
12. John CD, Buckingham JC. Cytokines: regulation of the hypothalamo-pituitary-adrenocortical axis. Curr Opinion Pharmacol. 2003;3:78-84.
13. Cooper MS, Stewart PM. 11 beta-hydroxysteroid dehydrogenase type 1 and its role in the hypothalamus-pituitary-adrenal axis, metabolic syndrome, and inflammation. J Clin Endocrinol Metab. 2009;94:4645-54.
14. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci. 2013;34:518-30.
15. Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111-33.
16. Savory JG, Hsu B, Laquian IR, Giffin W, Reich T, Hache RJ, Lefebvre YA. Discrimination between NL1- and NL2-mediated nuclear localization of the glucocorticoid receptor. Mol Cell Biol. 1999;19:1025-37.
17. Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17(3):245-61.
18. Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306-60.
19. Barnes PJ. How corticosteroids control inflammation: Quintiles Prize Lecture 2005. Br J Pharmacol. 2006;148:245-54.
20. Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR. Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol. 2017;18:159-74.
21. Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324(5925):407-10.
22. Bain DL, Yang Q, Connaghan KD, Robblee JP, Miura MT, Degala GD, et al. Glucocorticoid receptor-DNA interactions: binding energetics are the primary determinant of sequence-specific transcriptional activity. J Mol Biol. 2012;422:18-32.
23. Uhlenhaut NH, Barish GD, Yu RT, Downes M, Karunasiri M, Liddle C, et al. Insights into negative regulation by the glucocorticoid receptor from genome-wide profiling of inflammatory cistromes. Molecular Cell. 2013;49:158-71.
24. Liberman AC, Druker J, Perone MJ, Arzt E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine Growth Factor Rev. 2007;18:45-56.
25. Meijsing SH, Elbi C, Luecke HF, Hager GL, Yamamoto KR. The ligand binding domain controls glucocorticoid receptor dynamics independent of ligand release. Mol Cell Biol. 2007;27:2442-51.
26. Smoak KA, Cidlowski JA. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev. 2004;125:697-706.
27. De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene regression. Endocrine Rev. 2003;24:488-522.
28. Liberman AC, Druker J, Perone MJ, Arzt E. Glucocorticoids in the regulation of transcription factors that control cytokine synthesis. Cytokine & Growth Factor Rev. 2007;18:45-56.
29. Roth SY, Denu JM, Allis CD. Histone acetyltransferases. Annu Rev Biochem. 2001;70:81-120.
30. Ito K, Jazrawi E, Cosio B, Barnes PJ, Adcock IM. p65-activated histone acetyltransferase activity is repressed by glucocorticoids: Mifepristone fials to recruit HDAC2 to the 65/HAT complex. J Biol Chem. 2001;276:30208-15.
31. Ellmeier W, Seiser C. Histone deacetylase function in CD4+ T cells. Nat Rev Immunol. 2018 Jul 18. doi: 10.1038/s41577-018-0037-z.
32. Ganesan A. Epigenetic drug discovery: a success story for cofactor interference. Philos Trans R Soc Lond B Biol Sci. 2018;373(1748). pii: 20170069.
33. Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and coregulates major cellular functions. Science. 2009;325:834-40.
34. Dekker FJ, van den Bosch T, Martin NI. Small molecule inhibitors of histone acetyltransferases and deacetylases are potential drugs for inflammatory diseases. Drug Discov Today. 2014;19:654-60.
35. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110(1):93-105.
36. Stellato C. Post-transcriptional and nongenom¬ic effects of glucocorticoids. Proc Am Thorac Soc. 2004;1:255-63.
37. Hammes SR, Davis PJ. Overlapping nongenomic and genomic actions of thyroid hormone and steroids. Best Pract Res Clin Endocrinol Metab. 2015;29(4):581-93.
38. Ruhs S, Nolze A, Hübschmann R, Grossmann C. Nongenomic effects via the mineralocorticoid receptor. J Endocrinol. 2017;234:T107-T124.
39. Besedovsky H, del Rey A, Sorkin E, Dinarello CA. Immunoregulatory feedback between interleukin-1 and glucocorticoid hormones. Science. 1986;233:652-4.
40. Dunn AJ. Cytokine activation of the HPA axis. Ann NY Acad Sci. 2000;917:608-17.
41. Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M. Immunosupression by glucocorticoids: inhibition of NF-kB activity through induction of 1kappa B synthesis. Science. 1995;270:286-90.
42. De Bosscher K, Vanden Berghe W, Haegeman G. Mechanisms of antiinflammatory action and of immunosuppression by glucocorticoids: negative interference of activated glucocorticoid receptor with transcription factors. J Neuroimmunol. 2000;109:16-22.
43. Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol. 2009;39:3147-59.
44. Rea D, van Kooten C, van Meijgaarden KE, Ottenhoff TH, Melief CJ, Offringa R. Glucocorticoids transform CD-40-triggering of dendritic cells into an alternative activation pathway resulting in antigenpresenting cells that secrete IL-10. Blood. 2000;95:3162-7.
45. Varga G, Ehrchen J, Brockhausen A, Weinhage T, Nippe N, Belz M, et al. Immune suppression via glucocorticoid-stimulated monocytes: a novel mechanism to cope with inflammation. J Immunol. 2014;193:1090-9.
46. Giembycz MA, Lindsay MA. Pharmacology of the eosinophil. Prharmacol Rev. 1999;51:213-340.
47. Hallett JM, Leitch AE, Riley NA, Duffin R, Haslett C, Rossi AG. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol Sci. 2008;20:250-7.
48. Donnelly LE, Barnes PJ. Defective phagocytosis in airway disease. Chest. 2012;141:1055-62.
49. Umland SP, Schleimer RP, Johnston SL. Review of the molecular and cellular mechanisms of action of glucocorticoids for use in asthma. Pulm Pharmacol Ther. 2002;15:35-50.
50. Rhen T, Cidlowski JÁ. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med. 2005;353:1711-23.
51. Durham AL, Caramori G, Chung KF, Adcock IM. Targeted antiinflammatory therapeutics in asthma and chronic obstructive lung disease. Transl Res. 2016;167(1):192-203.
52. Duong-Thi-Ly H, Nguyen-Thi-Thu H, Nguyen-Hoang L, Nguyen- Thi-Bich H, Craig TJ, Duong-Quy S. Effects of genetic factors to inhaled corticosteroid response in children with asthma: a literature review. J Int Med Res. 2017;45(6):1818-30.
53. Newton R, Giembycz MA. Understanding how long-acting ß2-adrenoceptor agonists enhance the clinical efficacy of inhaled corticosteroids in asthma - an update. Br J Pharmacol. 2016;173(24):3405-30.
54. Williamson IJ, Matusiewicz SP, Brown PH, Greening AP, Crompton GK. Frequency of voice problems and cough in patients using pressurized aerosol inhaled steroid preparations. Eur Respir J. 1995;8(4):590.
55. Yang IA, Fong KM, Sim EH, Black PN, Lasserson TJ. Inhaled corticosteroids for stable chronic obstructive pulmonary disease. Cochrane Database Syst Rev. 2012.
56. Lipworth BJ. Systemic adverse effects of inhaled corticosteroid therapy: A systematic review and meta-analysis. Arch Intern Med. 1999;159(9):941.
57. Patel L, Wales JK, Kibirige MS, Massarano AA, Couriel JM, Clayton PE. Symptomatic adrenal insufficiency during inhaled corticosteroid treatment. Arch Dis Child. 2001 Oct;85(4):330-4.
58. Cazeiro C, Silva C, Mayer S, Mariany V, Wainwright CE, Zhang L. Inhaled Corticosteroids and respiratory infections in children with asthma: a meta-analysis. Pediatrics. 2017;139(3). pii: e20163271.
59. van der Wiel E, ten Hacken NH, Postma DS, van den Berge M. Small-airways dysfunction associates with respiratory symptoms and clinical features of asthma: A systematic review. J Allergy Clin Immunol. 2013;131:646-57.
60. Johal B, Howald M, Fischer M, Marshall J, Venthoye G. Fine particle profile of fluticasone propionate/formoterol fumarate versus other combination products: the DIFFUSE study. Comb Prod Ther. 2013;3:39-51.
61. Brown J, Zeman K, Bennet W. Ultrafine particle deposition in the healthy and obstructed lung. Am J Crit Care Med. 2002;166:1240-7.
62. El Baou C, Di Santostefano RL, Alfonso-Cristancho R, Suarez EA, Stempel D, Everard ML, et al. Effect of inhaled corticosteroid particle size on asthma efficacy and safety outcomes: a systematic literature review and meta-analysis. BMC Pulm Med. 2017;17(1):31.
63. Leach CL, Davidson PJ, Hasselquist BE, Boudreau RJ. Lung deposition of hydrofluoroalkane-134a beclomethasone is greater than that of chlorofluorocarbon fluticasone and chlorofluorocarbon beclomethasone: a cross-over study in healthy volunteers. Chest. 2002;122:510-6.
64. Carvalho TC, Peters JI, Williams RO 3rd. Influence of particle size on regional lung deposition – what evidence is there? Int J Pharm. 2011;406:1-10.
65. Johnson M. Pharmacodynamics and pharmacokinetics of inhaled glucocorticoids. J Allergy Clin Immunol. 1996;97(1 Pt 2):169.
66. Czock D, Keller F, Rasche FM, Häussler U. Pharmacokinetics and pharmacodynamics of systemically administered glucocorticoids. Clin Pharmacokinet. 2005;44(1):61-98.
67. Jiang CL, Liu L, Li Z, Buttgereit F. The novel strategy of glucocorticoid drug development via targeting nongenomic mechanisms. Steroids. 2015;102:27-31.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888