Número Atual: Abril-Junho 2018 - Volume 2 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Síndrome de Chediak-Higashi em fase acelerada: um relato de caso
Accelerated Chediak-Higashi syndrome: case report
Aline Mendes; Constantino Giovanni Braga Cartaxo
Centro de Ciências Médicas - Universidade Federal da Paraíba, Joao Pessoa, PB, Brasil
Endereço para correspondência:
Aline Mendes
E-mail: li.m@hotmail.com
Submetido em 19/02/2018
Aceito em: 10/03/2018
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
A síndrome de Chediak-Higashi (CHS) é um distúrbio genético autossômico recessivo decorrente de uma mutaçao no gene regulador do transporte lisossomal (LYST ou CHS1). Os sintomas da síndrome sao resultado de alteraçoes funcionais de melanócitos, plaquetas, neutrófilos e células natural killer, e incluem albinismo parcial, fotossensibilidade, infecçoes recorrentes, principalmente bacterianas, linfocitose hemofagocítica, sangramentos e manifestaçoes neurológicas, como neuropatia central e periférica, perda de sensibilidade, fraqueza muscular, ataxia cerebelar e déficit cognitivo. Aproximadamente 85% dos casos se apresentam como a forma avançada, caracterizada por pancitopenia, hemofagocitose e infiltrado linfocítico em todos os órgaos, determinando falência múltipla dos órgaos. Nesse estudo é relatado o caso de uma paciente diagnosticada com a síndrome aos 8 anos de idade, apresentando a doença já em fase avançada, além de uma rápida revisao bibliográfica sobre a doença em questao.
Descritores: Chediak-Higashi, linfohistiocitose hemofagocítica, relato de caso.
INTRODUÇAO
A síndrome de Chediak-Higashi (CHS) é um distúrbio genético autossômico recessivo, decorrente de mutaçao no gene regulador do transporte lisossomal (LYST ou CHS1)1,2. Os sintomas da síndrome sao resultado de alteraçoes funcionais de melanócitos, plaquetas, neutrófilos e da funçao das células NK, e incluem albinismo parcial, fotossensibilidade, infecçoes recorrentes, principalmente bacterianas, linfocitose hemofagocítica, sangramentos e manifestaçoes neurológicas, como neuropatia central e periférica, perda de sensibilidade, fraqueza muscular, ataxia cerebelar e déficit cognitivo3. Nos últimos 20 anos, menos de 500 casos da doença foram descritos em todo o mundo4. Por volta de 50 a 85% dos casos evoluem para a forma avançada da doença, também chamada de linfo-histiocitose hemofagocítica, caracterizada por pancitopenia, hemofagocitose e infiltrado linfocítico em todos os órgaos, podendo levar à falência múltipla dos órgaos5.
Nesse estudo, é relatado o caso de um paciente diagnosticada aos 7 anos de idade em fase avançada da Síndrome de Chediaki-Higashi, com o objetivo de chamar atençao para uma doença potencialmente fatal e ainda pouco conhecida na prática médica.
RELATO DE CASO
Paciente R.C.S.S., sexo feminino, 8 anos de idade, admitida no hospital devido à quadro de astenia, febre e lesoes periorais, sendo entao diagnosticada com Herpes simples. É filha única de pais consanguíneos, sem antecedentes patológicos e com desenvolvimento neuropsicomotor normal. Durante a internaçao evoluiu com colúria, hipocolia fecal e elevaçao de enzimas hepáticas. Foi tratada com Aciclovir e Cefotaxima por nove dias, com melhora parcial dos sintomas e recebendo alta hospitalar.
Vinte dias após a alta foi reinternada com quadro de febre, astenia, desconforto respiratório e um episódio isolado de epistaxe. Apresentava-se ictérica, com nistagmo rotatório bilateral, hepatoesplenomegalia, albinismo e cabelo com coloraçao acinzentada. Os exames laboratoriais evidenciaram pancitopenia, hiperferritinemia (> 40.000 µg/L), hipofibrinogenemia e hipertrigliceridemia, e o esfregaço de sangue periférico mostrou a presença de leucócitos com grânulos gigantes em seu citoplasma. Foi coletado mielograma no dia seguinte da admissao, sendo visualizado grânulos azurófilos gigantes (Figura 1) que, juntamente com o quadro clínico, confirmou o diagnóstico de Chediaki-Higashi com linfohistiocitose hemofagocítica.
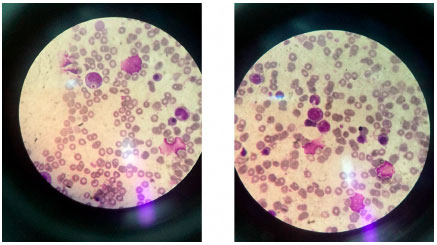
Figura 1 Aspirado de medula óssea evidenciando grânulos azurófilos gigantes
A paciente evoluiu com infecçoes secundárias, tratada com antibióticos e antifúngicos, além de suporte hematológico com transfusoes de concentrados de plaquetas e de hemácias.
Os sinais, sintomas e achados laboratoriais de reativaçao macrofágica ocorreram por quatro vezes durante a internaçao, com melhora com o uso de Ciclosporina e Dexametasona. Em um destes episódios a paciente nao apresentou melhora, sendo entao associado o uso de imunoglobulina na dose de 400 mg/kg/dia por 5 dias, havendo melhora do quadro clinico e dos parâmetros laboratoriais, com diminuiçao da hepatoesplenomegalia. Manteve-se entao com o uso semanal de imunoglobulina e pulsoterapias com metilprednisolona nas recorrências clínica e laboratoriais.
Após seis meses de internaçao foi transferida para outro serviço para a realizaçao de transplante de medula óssea, inicialmente sem intercorrências, porém com óbito uma semana após o procedimento.
DISCUSSAO
O primeiro registro da Síndrome de Chediak-Higashi (CHS) é datado de 1943, feito por Beguez-Cesar. Nove anos depois, Moisés Chediak descreveu quatro casos da doença em 13 irmaos, e Ototaka Higashi, em 1954, relatou a mesma doença em irmaos japoneses, sendo o epônimo Síndrome de Chediak-Higashi usado pela primeira vez em 19556.
A CHS é um distúrbio genético autossômico recessivo raro, com aproximadamente 500 casos descritos no mundo até os dias de hoje7, que surge a partir de uma mutaçao no gene regulador da proteína do transporte lisossomal (LYST ou CHS1), localizado no cromossomo 11,2. Essa proteína tem como funçao a síntese, transporte e fusao das vesículas citoplasmáticas, impedindo a incorporaçao inadequada de proteínas na membrana lisossomal. Sendo assim, a mutaçao no gene em questao leva à alteraçao no funcionamento, morfologia e localizaçao das vesículas/grânulos do citoplasma de diversas células, principalmente dos neutrófilos polimorfonucleares, melanócitos e células natural killer (NK)7,8. Essas alteraçoes, vistas à análise da medula óssea ou análise de sangue periférico, se apresentam como grânulos azurófilos gigantes, que é sinal patognomonico da CHS.
A idade média de diagnóstico é de 5 anos, sendo que a maioria dos pacientes morrem durante a primeira década de vida7, devido, principalmente, à infecçoes, sangramentos ou síndrome hemofagocítica. Consanguinidade dos pais está presente em 50% dos casos, como no caso aqui apresentado.
A CHS se caracteriza por albinismo parcial (graus variados de hipopigmentaçao em pele, olhos e cabelo), fotossensibilidade, infecçoes recorrentes, principalmente bacterianas, linfocitose hemofagocítica, sangramentos e manifestaçoes neurológicas, que variam de neuropatia central e periférica, perda de sensibilidade, fraqueza muscular, ataxia cerebelar à déficit cognitivo. Essas alteraçoes sao resultado da disfunçao funcional de melanócitos, plaquetas, neutrófilos e da funçao das células NK.
Sua classificaçao é baseada na idade em que a doença se apresenta: durante a infância (precoce), ou na adolescência/durante a fase adulta (tardia). A forma tardia, encontrada em pacientes durante a adolescência ou a vida adulta, costuma se apresentar de forma menos agressiva clinicamente, sendo que esses pacientes evoluem com poucas infecçoes durante os primeiros anos de vida. Porém, nesses casos, os sintomas neurológicos sao mais expressivos, apresentando-se com déficit cognitivo, neuropatias e paresias. Cerca de 80% dos casos apresenta-se com manifestaçoes clínicas durante a infância, com infecçoes recorrentes, principalmente de vias aéreas, e evoluçao para a fase acelerada da doença.
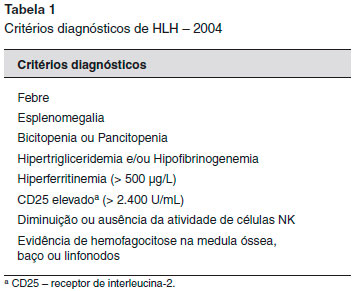
A fase acelerada da CHS, chamada de linfohistiocitose hemofagocítica (HLH) ou síndrome hemofagocítica primária ou familiar, ocorre em 50 a 85% dos casos, sendo letal se nao tratada4. A HLH é caracterizada por uma reaçao inflamatória intensa e generalizada, que se inicia com ativaçao excessiva de linfócitos T CD8 e histiócitos, com consequente aumento na produçao de citocinas como o fator de necrose tumoral alfa (TNF-α), Interferon gama (IFN-β), interleucinas e CD-25. O TNF-α contribui para a ativaçao de macrófagos, levando à hemofagocitose e consequente citopenia, e inibe a lipase de lipoproteínas, aumentando os níveis de triglicerídeos. Além disso, juntamente com o IFN-β e a interleucina 1β, o TNF-α diminui a hematopoiese, aumentando a citopenia. Os macrófagos ativados fagocitam outras células hematopoiéticas, além de secretarem ativador de plasminogênio e ferritina9. Para o diagnóstico, é necessário que o paciente apresente ao menos 5 dos 8 critérios estabelecidos pela Histiocyte Society no guideline de 2004, conforme demonstrado na Tabela 1.
Alguns pacientes nao apresentam achados suficientes para o diagnóstico definitivo, mesmo com grande suspeita da doença. Nesses casos, deve-se iniciar o tratamento precocemente, antes que os danos causados pela atividade da doença se tornem irreversíveis e letais. Outros achados que corroboram o diagnóstico sao: icterícia, alteraçao de enzimas hepáticas, aumento de lactato desidrogenase, hipoproteinemia, derrame pleural ou pericárdico, hiponatremia, linfonodomegalias, aumento de VLDL, diminuiçao de HDL, edema, rash cutâneo e sintomas neurológicos4,10.
Sem a correta terapia para o controle da linfohistiocitose hemofagocítica, todos os pacientes evoluem para o óbito. Para nortear o tratamento, foi realizado um estudo terapêutico pela Histiocyte Society denominado HLH-2004, que tinha como objetivo aumentar a taxa de sobrevida das crianças afetadas pela HLH e divide-se em duas fases, em que a fase inicial visa diminuir a fase ativa da doença, levando-a à remissao, e a segunda fase (de manutençao) tem como objetivo manter o paciente livre de infecçoes e em condiçoes para realizar o transplante de medula óssea, padrao ouro para o tratamento de HLH familiar. O principal objetivo desse tratamento é diminuir o intenso processo inflamatório determinante da doença.
A primeira fase possui duraçao de 8 semanas, onde é realizado:
1. Etoposídeo: 150 mg/m2 de superfície corpórea duas vezes na semana por 2 semanas, seguido por uso semanal por 6 semanas;
2. Dexametasona: 10 mg/m2 por 2 semanas, seguido de uma reduçao de 50% na dose a cada 2 semanas, até sua retirada completa após 8 semanas;
3. Ciclosporina A: 6 mg/kg/dia dividido em duas doses, visando manter níveis séricos de 200 µg/L
A fase de manutençao, realizada a partir da nona semana, consiste em pulsoterapias com dexametasona na dose de 10 mg/m2 por 3 dias a cada duas semanas, alternando com etoposídeo 150 mg/m2 a cada duas semanas e Ciclosporina A11. O uso de metotrexato está indicado apenas se há sintomas neurológicos, no uso máximo de 4 doses. Além disso, é recomendado o uso de antibioticoprofilaxia quando em uso de dexametasona, a fim de diminuir o risco de novas infecçoes.
Mesmo com o seguimento do protocolo adequadamente, a possibilidade de ocorrer reicidivas com reativaçao da doença é grande. Nesses casos, o tratamento deve ser individualizado conforme cada paciente, com base na sua melhora clínica e laboratorial frente ao uso das medicaçoes.
O tratamento definitivo da Síndrome de Chediak-Higashi se dá somente por meio do transplante de medula óssea, sendo que o momento ideal para a realizaçao do mesmo é logo ao diagnóstico, antes do paciente apresentar a doença em fase avançada. Contudo, mesmo após o transplante é possível que haja progressao dos sintomas neurológicos.
CONCLUSAO
A Síndrome de Chediak-Higashi é uma enfermidade rara e de evoluçao grave, se nao diagnosticada de forma correta. Aproximadamente 80% dos pacientes diagnosticados se encontram na primeira infância, fase em que a doença se apresenta, em geral, de forma mais agressiva, podendo evoluir rapidamente para a fase acelerada. Se constatada a linfohistiocitose hemofagocítica, se faz necessário uma intervençao rápida, seguindo o protocolo criado em 2004 pela Histiocyte Society. Contudo, vale ressaltar que a resposta do paciente às medicaçoes utilizadas é o parâmetro mais importante para ajuste do tratamento.
Quando a doença é diagnosticada, é necessário um acompanhamento multidisciplinar do paciente, evitando infecçoes afim de mantê-lo estável clinicamente até a realizaçao do transplante de medula óssea.
REFERENCIAS
1. Gil-Krzewska A, Wood SM, Murakami Y, Nguyen V, Chiang SC, Cullinane AR, et al. Chediak-Higashi syndrome: Lysosomal trafficking regulator domains regulate exocytosis of lytic granules but not cytokine secretion by natural killer cells. J Allergy Clin Immunol. 2016;137(4):1165-77.
2. Shashikant C, Kumar S, Bagri N, Kumar A, Shukla J. Chediak- Higashi Syndrome: a case report. Indian Journal of Hematology & Blood Transfusion. 2013;29(2):80-3.
3. Proverbio T, Proverbio F, Marin R, Merino F. Na K-ATPase activity in red blood cells from patients with Chediak-Higashi Syndrome. Experimental and Molecular Pathology. 1995;62:173-9.
4. Wu XL, Zhao XQ, Zhang BX, Xuan F, Guo HM, Ma FT. A novel frameshift mutation of Chediak-Higashi syndrome and treatment in the accelerated phase. Braz J Med Biol Res. 2017 Mar 23;50(4):e5727.
5. Carnide E, Jacob C, Pastorino A, Bellinati-Pires R, Costa M, Grumach A. Chédiak-Higashi syndrome: presentation of seven cases. Sao Paulo Medical Journal. 1998;116(6):1873-18.
6. Adams R, et al. Neurocutaneous Diseases: a practical approach. Boston: Butterworth Publishers; 1987.
7. Maalouol I, Talmoudi J, Chabchoub I, Ayadi L, Kamoun TH, Boudawara T, et al. Chediak-Higashi syndrome presenting in accelerated phase: a case report and literature review. Hematology. Hematology/Oncology and Stem Cell Therapy. 2016;9(2):71-5.
8. Ortuno F, Fuster J, Jerez A. Sindrome de Chediak-Higashi. Med Clin (Barc). 2010;135(11):512-8.
9. Jobim M, Trotta E, Pilcher O, Fernandes FB, Daut L, Jobim LF. Linfo-histiocitose hemofagocítica: tratamento com plasmaferese e gamaglobulina endovenosa. Revista da AMRIGS. 2010;54(1):72-6.
10. Henter JI, Horne A, Aricó M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood Cancer. 2007;48( 2):124-31.
11. Henter JI, Samuelsson-Horne A, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002;100(7):2367-73.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888