Número Atual: Abril-Junho 2017 - Volume 1 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicação Clínica e Experimental
Doença sistêmica relacionada à IgG4 com linfopenia: relato de caso e breve revisão de literatura
IgG4-related systemic disease associated with lymphopenia: case report and brief literature review
Leonardo Oliveira Mendonça; Julia Biegelmeyer; Joao Paulo de Assis; Larissa Costa Amorim; Mauricio Levy-Neto; Jorge Kalil; Milton de Arruda Martins; Fabio Morato Castro; Myrthes Toledo de Barros
DOI: 10.5935/2526-5393.20170028
Disciplina de Imunologia Clínica e Alergia da Faculdade de Medicina da Universidade de Sao Paulo, Sao Paulo, SP
Endereço para correspondência:
Leonardo Oliveira Mendonça
E-mail: leonardo.oliveira.mendonca@gmail.com
Submetido em: 10/04/2017
Aceito em: 24/04/2017.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
A doença sistêmica relacionada à IgG4 é uma doença emergente, recentemente descrita, caracterizada clinicamente por aumento parcial ou total de um órgao, e, por isso, com amplo espectro de manifestaçoes clínicas. Esta é uma doença sistêmica fibroinflamatória, patologicamente provocada pela infiltraçao de plasmoblastos IgG4 positivos que levam à inflamaçao eosinofílica do tecido e, consequentemente, fibrose estoriforme. Quando o diagnóstico é precoce, a melhora clínica e resposta sustentada com corticosteroides sistêmicos é impressionante. O diagnóstico é baseado em critérios patológicos, mas, recentemente, alguns trabalhos têm descrito que plasmoblastos no soro podem servir como um fator independente para auxiliar no diagnóstico da doença. Este artigo descreve uma apresentaçao atípica da doença relacionada à IgG4, em um paciente linfopênico com mediçao inconclusiva de plasmoblastos no soro.
Descritores: Imunoglobulina G, eosinófilos, hipergamaglobulinemia, imunoglobulinas, serosite, IgG4, doença sistêmica relacionada à IgG4.
RESUMO
A doença sistêmica relacionada à IgG4 é uma doença emergente, recentemente descrita, caracterizada clinicamente por aumento parcial ou total de um órgao, e, por isso, com amplo espectro de manifestaçoes clínicas. Esta é uma doença sistêmica fibroinflamatória, patologicamente provocada pela infiltraçao de plasmoblastos IgG4 positivos que levam à inflamaçao eosinofílica do tecido e, consequentemente, fibrose estoriforme. Quando o diagnóstico é precoce, a melhora clínica e resposta sustentada com corticosteroides sistêmicos é impressionante. O diagnóstico é baseado em critérios patológicos, mas, recentemente, alguns trabalhos têm descrito que plasmoblastos no soro podem servir como um fator independente para auxiliar no diagnóstico da doença. Este artigo descreve uma apresentaçao atípica da doença relacionada à IgG4, em um paciente linfopênico com mediçao inconclusiva de plasmoblastos no soro.
Descritores: Imunoglobulina G, eosinófilos, hipergamaglobulinemia, imunoglobulinas, serosite, IgG4, doença sistêmica relacionada à IgG4.
ABSTRACT
IgG4-related systemic disease is a recently described, emerging condition, clinically characterized by partial or total enlargement of an organ, with a broad spectrum of clinical manifestations. It is a systemic fibroinflammatory condition caused by the infiltration of IgG4-positive plasmablasts that lead to eosinophilic inflammation of tissues and consequently storiform fibrosis. When diagnosis is early, clinical improvement and maintained response achieved with systemic corticosteroids is impressive. Diagnosis is based on pathological criteria, but recent papers have described that serum plasmablasts may serve as an independent factor to aid in diagnosis. This paper describes an atypical presentation of IgG4-related disease in a lymphopenic patient with inconclusive serum plasmablast measurement.
Keywords: Immunoglobulin G, eosinophils, hypergammaglobulinemia, immunoglobulins, serositis, IgG4, IgG-4 related systemic disease.
INTRODUÇAO
A doença sistêmica relacionada à IgG4 (DRIgG4) resulta de funçao anormal da molécula de IgG4, que normalmente corresponde a 5% (< 140 mg/dL) de todas imunoglobulinas do soro humano1. A funçao fisiológica da IgG4 é auxiliar o controle da resposta Th-2, através de reversao do pool de citocinas 4 e 13 (IL-4 e IL-13) em 10 (IL-10), a, assim chamada, resposta Th-2 modificada2. Estudos in vitro demonstraram que a IgG4 nao ativa a via clássica do complemento, e que nao tem um papel importante na ativaçao da resposta imune adaptativa através do componente Fc da imunoglobulina3. A IgG4 patológica implica em atividade de fator reumatoide, com a ativaçao do complemento e atividade anormal Fc que desencadeia danos nos tecidos4.
As manifestaçoes clínicas estao normalmente relacionadas com tumefaçao dos órgaos e fibrose. Ela pode se manifestar em um único órgao ou difusamente. Anormalidades laboratoriais sao níveis elevados de imunoglobulina E (IgE), eosinófilos séricos e IgG4, embora níveis normais de IgG4 sao observados em 20-30% dos pacientes5,6. Elevaçao das provas de fase aguda como PCR, VHS e níveis de ferritina sao observados em alguns pacientes. Alguns pacientes apresentam positividade de fator reumatoide e consumo da fraçao C4 do complemento7. O padrao ouro e a confirmaçao do diagnóstico é realizada pela biópsia do órgao afetado, com critérios patológicos bem estabelecidos8,9. Recentemente, demonstrou-se que a concentraçao de plasmoblastos ativados sanguíneos pode ser superior à dosagem de IgG4 no diagnóstico da doença8. A mensuraçao de plasmoblastos por mililitro no sangue periférico é realizada por citometria de fluxo com o painel CD19+, CD20-, CD38+ e CD27+.
Desde que a DRIgG4 tem sido relatada, foi observado um espectro amplo de manifestaçoes clínicas. Este relato de caso tem como objetivo descrever uma apresentaçao clínica interessante, difícil e rara da DRIgG4, e discutir seu diagnóstico.
RELATO DE CASO
ZMS, 63 anos de idade, branca, do sexo feminino, casada, nascida em Sao Paulo, cresceu em Itajubá, Minas Gerais, Brasil, com diagnóstico prévio de diabetes mellitus tipo 2 há um ano. A paciente foi admitida pela primeira vez em sua cidade natal com história de 30 dias de perda de peso e dispneia. Naquele momento teve diagnóstico de derrame pleural e pericárdico, que foram drenados e eram compatíveis com exsudato. Recebeu alta com resultados inconclusivos.
Depois de um mês, ainda com derrame pleural, começou a apresentar aumento do volume abdominal. Readmitida em enfermaria de um hospital quaternário e submetida à tomografia computadorizada (TC) abdominal que revelou ascite volumosa, aumento difuso do pâncreas, dilataçao do colédoco e estenose da porçao cefálica do pâncreas, o que sugeria pancreatite aguda. A paciente nao relatava dor abdominal e nao apresentava níveis de lipase e amilase elevados. Afim de melhor avaliar os dutos biliares e pancreático, foi realizada uma colangiografia que confirmou os achados da TC. A paciente foi tratada com furosemida e espironolactona, com melhora clínica temporária.
Quatro meses depois, ela foi admitida com os mesmos sintomas em nosso hospital para tratamento e elucidaçao diagnóstica, com a principal hipótese diagnóstica de lupus eritematoso sistêmico. Apresentava poliserosite, com derrame pleural e pericárdico compatíveis com exsudato e ascite com um gradiente soro-ascite de albumina (SAAG) de 1,1 (Tabela 1). As funçoes renal e hepática eram normais, mas o nível de albumina sérica era de 2,4. Nao havia evidências de hepatite ou infecçao por HIV. Investigaçao imunológica revelou FAN padrao pontilhado fino em títulos de 1:320 e níveis de IgG normais. Apresentava anemia por deficiência de ferro e vitamina B12 baixa.
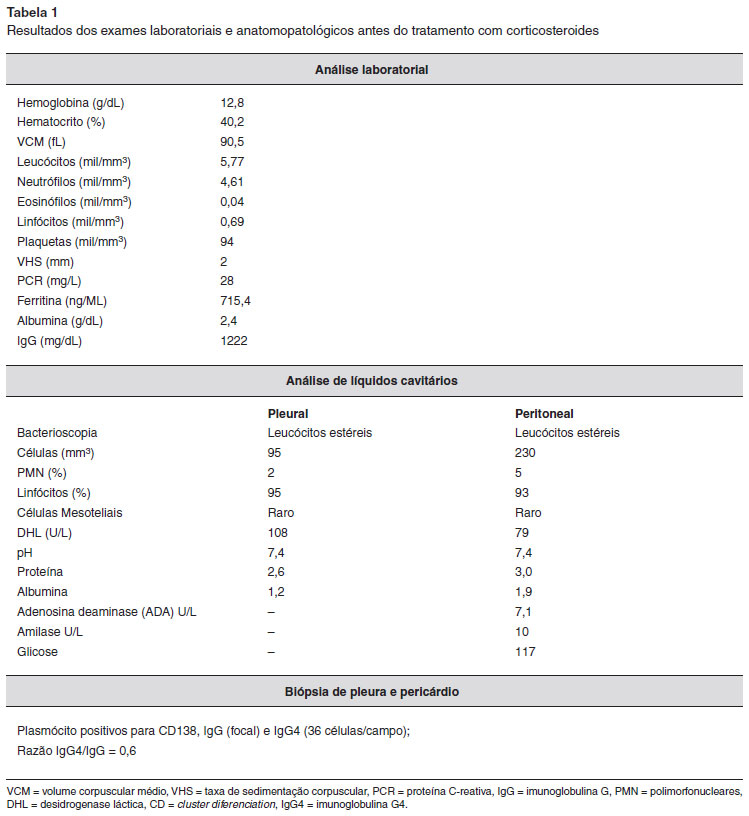
Uma vez que a paciente precisava de paracenteses semanais, decidiu-se começar um tratamento empírico com a dose imunossupressora de prednisona (1 mg/kg/dia). Foi entao solicitado análise de plasmoblastos do sangue periférico, a qual foi inconclusiva. A reavaliaçao da biópsia de pleura e pericárdio realizadas no início do quadro verificou infiltrado linfoplasmocitário, com expressao de CD138, IgG e IgG4, e relaçao IgG4/IgG de 0,6. A paciente recebeu alta e durante acompanhamento ambulatorial apresentou melhora da inflamaçao (PCR de 28 mg/L foi para 6,4 mg/L após tratamento) e normalizaçao dos níveis de albumina, com diminuiçao progressiva da necessidade de paracentese. Manteve uso de prednisona 60 mg/dia.
DISCUSSAO
A paciente aqui relatada apresentou pancreatite aguda, manifestaçao típica da DRIgG4, mas com evoluçao clínica e análise laboratorial inesperadas. As manifestaçoes clínicas descritas desta síndrome relativamente nova decorrem da disfunçao, com aumento de volume, de um único ou de múltiplos órgaos, o que ocorre em 60-90% dos casos10-16 (Tabela 2). As características clínicas usuais sao inespecíficas e compreendem linfadenopatia, sintomas de alergia ou de asma (40% dos pacientes), perda de peso e ausência de febre17. A DRIgG4 pode também simular outras doenças autoimunes, como lúpus eritematoso sistêmico, síndrome de Sjögren ou granulomatose com poliangeítes18. Algumas publicaçoes recentes descreveram os derrames pericárdico e pleural como manifestaçoes desta síndrome19-22. A atopia é tida como fator associado à DRIgG, mas somente é observada em alguns pacientes. Em uma casuística de 70 pacientes com doença relacionada à IgG4, apenas 22 deles eram atópicos, dos quais 73% apresentavam rinite alérgica, a doença mais prevalente23.

O diagnóstico da DRIgG4 requer quadro clínico compatível como descrito acima e pelo menos um marcador de laboratório (IgG4 sérica, plasmoblastos sanguíneos ativados, ou reagentes de fase aguda), além dos critérios patológicos. Pacientes que têm infiltraçao de mais de um órgao e/ou quadros agressivos apresentam elevaçao dos marcadores de fase aguda, como o VHS e o PCR, que sao os principais marcadores de atividade ou recaída da doença1,5,24,25. Nossa paciente apresentava elevaçao da PCR, que apresentou reduçao após a introduçao da corticoterapia. As dosagens de eosinófilos sanguíneos e IgE sérica estao geralmente normais na DRIgG4, com exceçao dos paciente que têm doenças atópicas associadas23.
A dosagem de IgG4 está elevada em apenas 51-61% dos casos, sendo maior em pacientes atópicos, e o seu valor normal nao exclui a doença1,5,23. Apenas as evidências clínicas e altos níveis de IgG4 sérica também nao sao suficientes para confirmar o diagnóstico, porque outras condiçoes podem elevar os títulos de IgG4. Há aumento policlonal das demais imunoglobulinas em aproximadamente 50% dos pacientes5. Recentemente, a pesquisa de plasmoblastos em sangue periférico mostrou-se um bom biomarcador para o diagnóstico, atividade da doença e avaliaçao da resposta terapêutica, indepentende da medida de IgG48, podendo inclusive ser melhor do que a dosagem sérica da IgG4. A análise de plasmoblastos do sangue periférico foi inconclusiva no caso presente.
O padrao ouro e o único teste diagnóstico confirmatório para DRIgG4 é a biópsia de tecido, que também tem a funçao de excluir malignidade27. Os critérios patológicos sao claros e é necessário pelo menos dois deles para confirmar o diagnóstico da doença28:
- Infiltrado linfoplasmocitário;
- Fibrose estoriforme;
- Flebite obliterante;
- Infiltrado eosinofílico;
- Presença de IgG4.
As biópsias da pleura e pericárdio associadas à resposta terapêutica aos glicocorticoides confirmaram o diagnóstico de DRIgG4 em nossa paciente.
A DRIgG4 é ainda de difícil reconhecimento na prática clínica, mas sempre tem que ser considerada como um diagnóstico diferencial em condiçoes fibroinflamatórias. A mensuraçao da IgG4 sérica e dos plasmoblastos sanguíneos nem sempre auxiliam no diagnóstico da doença.
REFERENCIAS
1. Engelhart S, Glynn RJ, Schur PH. Disease associations with isolated elevation of each of the four IgG subclasses. Semin Arthritis Rheum. 2017; no prelo.
2. Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539-51.
3. Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol. 2011;23:119-24.
4. Rispens T, Ooievaar-De Heer P, Vermeulen E, Schuurman J, Van der Neutkolfschoten M, Aalberse RC. Human IgG4 binds to IgG4 and conformationally altered IgG1 via Fc-Fc interactions. J Immunol. 2009;182:4275-81.
5. Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, Pillai S, Stone JH. IgG4-Related Disease: Clinical and Laboratory Features in One Hundred Twenty-Five Patients. Arthritis Rheumatol. 2015;67:2466-75.
6. Anchez-Castanon M, las Heras-Castano G, Lopez-Hoyos M. Autoimmune pancreatitis: an underdiagnosed autoimmune disease with clinical, imaging and serological features. Autoimmun Rev. 2010;9:237-40.
7. Kubo K, Yamamoto K. IgG4-related disease. Int J Rheum Dis. 2016;19:747-62.
8. Wallace ZS, Mattoo H, Carruthers M, Vinay MS, Emanuel DT, Lee H, et al. Plasmablasts as a biomarker for IgG4-related disease, independent of serum IgG4 concentrations. Ann Rheum Dis. 2015;74:190-5.
9. Deshpande V, Zen Y, Chan JKC, Yi EE, Sato Y, Yoshino T, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25:1181-92.
10. Thompson A, Whyte A. Imaging of IgG4-related disease of the head and neck. Clin Radiol. 2017;no prelo.
11. Narayan AK, Baer A, Fradin J. Sonographic findings of IgG4-related disease of the salivary glands: case report and review of the literature. J Clin Ultrasound. 2017;no prelo.
12. Khosroshahi A, Stone JH. A clinical overview of IgG4-related systemic disease. Curr Opin Rheumatol. 2011;23:57-66.
13. Takahashi H, Yamamoto M, Suzuki C, Naishiro Y, Shinomura Y, Imai K. The birthday of a new syndrome: IgG4-related diseases constitute a clinical entity. Autoimmun Rev. 2010;9:591-4.
14. Okazaki K, Uchida K, Koyabu M, Miyoshi H, Takaoka M. Recent advances in the concept and diagnosis of autoimmune pancreatitis and IgG4-related disease. J Gastroenterol. 2011;46:277-88.
15. Sah RP, Chari ST, Pannala R, Sugar A, Claim JE, Ley MJ, et al. Differences in clinical profile and relapse rate of type 1 versus type 2 autoimmune pancreatitis. Gastroenterology. 2010;139:140-8.
16. Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol. 2010;17:303-32
17. Soliotis F, Mavragani CP, Plastiras SC, Rontogianni D, Skopouli FN, Moutsopoulos HM. IgG4-related disease: a rheumatologist's perspective. Clin Exp Rheumatol. 2014;32:724-7.
18. Ishida A, Furuya N, Nishisaka T, Nishisaka T, Mineshita M, Miyazawa T. IgG4-related Pleural Disease Presenting as a Massive Bilateral Effusion. J Bronchology Interv Pulmunol. 2014;21:237-41.
19. Erlij D, Ramos D, Montana J, Kusnir P, Correa G, Neira O. IgG4- related disease, the new "great mimicker": Report of one case. Rev Med Chil. 2014;142:646-50.
20. Ishida M, Hodohara K, Furuya A, Fujishiro A, Okuno H, Yoshii M, et al. Concomitant occurrence of IgG4-related pleuritis and periaortitis: a case report with review of the literature. Int J Clin Exp Pathol. 2014;7:808-14.
21. Choi JH, Sim JK, Oh JY, Lee EJ, Hur GY, Lee SH, et al. A Case of IgG4-Related Disease Presenting as Massive Pleural Effusion and Thrombophlebitis. Tuberc Respir Dis (Seoul). 2014;76:179-83.
22. Rossi G, Marchioni A, Guicciardi N, Cadioli A, Cavazza A. Recurrent pleural and pericardium effusions in a white woman with IgG4-related syndrome. Am J Surg Pathol. 2009;33:802-3.
23. Della Torre E, Mattoo H, Mahajan VS, Carruthers M, Pillai S, Stone JH. Prevalence of Atopy, eosinophilia, and IgE elevation in IgG4- related disease. Allergy. 2014;69:269-72.
24. Hamano H, Kawa S, Horiuchi A, Unno H, Furuya N, Akamatsu T, et al. High serum IgG4 concentrations in patients with sclerosing pancreatitis. N Engl J Med. 2001;344:732-8.
25. Harano Y, Honda K, Akiyama Y, Kotajima L, Arioka H. A case of IgG4-Related Hypophysitis presented with Hypopituitarism and Diabetes Insipidus. Clin Med Insights Case Rep. 2015;8:23-6.
26. Mattoo H, Mahajan VS, Della-Torre E, Sekigami Y, Carruthers M, Wallace ZS, et al. De novo oligoclonal expansions of circulating plasmablasts in active and relapsing IgG4-related disease. J Allergy Clin Immunol. 2014;134:679-87.
27. Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34:1812-9.
28. Khosroshahi A, Wallace ZS, Crowe JL, Akamizu T, Azumi A, Carruthers MN, et al. International Consensus Guidance Statement on the Management and Treatment of IgG4-Related Disease. Arthritis Rheumatol. 2015;67:1688-99.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888