Número Atual: Abril-Junho 2017 - Volume 1 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Artigo de Revisão
O papel da IgG4 na fisiopatogenia da nefropatia membranosa idiopática: estado da arte
The role of IgG4 in the physiopathology of idiopathic membranous nephropathy: state of the art
Denise Maria do Nascimento Costa; Lucila Maria Valente; Gisélia Alves Pontes da Silva; Emanuel Sarinho
DOI: 10.5935/2526-5393.20170021
Universidade Federal de Pernambuco, UFPE, Recife, PE
Endereço para correspondência:
Denise Costa
E-mail: demnc@hotmail.com
Submetido em: 19/04/2017
Aceito em: 03/05/2017.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
Nefropatia membranosa idiopática é uma causa de síndrome nefrótica cuja etiopatogenia nao está completamente esclarecida. Trata-se de uma doença imunologicamente mediada, na qual a deposiçao de imunocomplexos decorre da reaçao antígenoanticorpo in situ, na regiao subepitelial glomerular. A maioria dos antígenos envolvidos identificados sao alvos da IgG4, subclasse predominante em imunofluorescências renais na nefropatia membranosa idiopática, em contraste com as formas secundárias da doença, nas quais IgG1, IgG2 e IgG3 prevalecem. Apesar da IgG4 ser um subtipo de imunoglobulina com baixa capacidade de ativaçao do complemento, há várias evidências deste envolvimento na glomerulopatia (GMP). Esses dados, em conjunto com achados de depósitos glomerulares de lectina ligadora de manose, um dos principais componentes da via das lectinas do complemento, podem sugerir que tanto a via da lectina como a IgG4 estao envolvidas nesta patologia. Os motivos que desencadeiam a formaçao dos imunocomplexos e a ativaçao das vias do complemento nesta doença sao incertos. A hipótese mais aceita é a de que a nefropatia membranosa idiopática resulte do conjunto de três condiçoes: presença de proteínas com conformaçoes alteradas que passam a atuar como autoantígenos, anticorpos do tipo IgG4 contra estes antígenos, e susceptibilidade genética. O objetivo foi verificar o possível papel da IgG4 na etiopatogenia da nefropatia membranosa primária segundo o que foi publicado até o momento na base de dados MEDLINE/PubMed, a partir de uma revisao narrativa.
Descritores: Glomerulonefrite membranosa, imunoglobulina G, ativaçao do complemento, etiologia.
INTRODUÇAO
A nefropatia membranosa (NM) é uma patologia cuja etiologia pode ser idiopática, ou ser associada a diversos formas de insultos. A nefropatia membranosa idiopática (NMI) teve seu modelo experimental inicial estabelecido em 1952, e, apesar dos avanços no seu entendimento, ainda nao tem mecanismos etiopatogênicos completamente elucidados. Provavelmente, o desenvolvimento de NMI está relacionado ao conjunto de três fatores: presença de auto antígenos podocitários, anticorpos do tipo IgG4 contra estes antígenos, e susceptibilidade genética1. Esta revisao narrativa delimita o conhecimento vigente a respeito dos mecanismos imunológicos relacionados à lesao glomerular na NMI.
A NEFROPATIA MEMBRANOSA
A NM é uma causa de síndrome nefrótica, caracterizada histopatologicamente por espessamento difuso de membrana basal glomerular, decorrente da deposiçao de complexos imunes na regiao podocitária. É classificada, de acordo com sua etiologia, como idiopática ou primária em cerca de 70 a 80% dos casos, quando nao há patologias subjacentes identificadas, ou secundária, podendo ser associada a causas autoimunes, infecciosas, neoplásicas e medicamentosas2. Dentre as formas secundárias, destaca-se o LES3, sendo a principal etiologia de NM secundária, principalmente em mulheres jovens4.
Apesar de rara, com incidência mundial de um caso a cada 100.000 indivíduos por ano, a NMI é uma das principais causas de síndrome nefrótica no mundo, responsável por cerca de 20% destes casos5, e é a segunda causa de síndrome nefrótica em adultos no Brasil6. Pode afetar indivíduos de todas as raças, sexos e idades, porém é mais comum em homens do que em mulheres (2:1), com pico de incidência entre os 30 e 50 anos de idade7.
A NMI é uma síndrome que pode resultar em grande morbidade e letalidade aos seus portadores. A síndrome nefrótica, presente em 60-80% dos pacientes, leva a aumento de complicaçoes cardiovasculares, incluindo eventos trombóticos (13%) e maior susceptibilidade a infecçoes (17%)8. A história natural desta doença é variável e imprevisível. Cerca de 30% dos pacientes apresentam remissao espontânea9 e outros 30% podem permanecer com proteinúria e com funçao renal estável. Porém, até 40% dos casos evoluem para doença renal crônica terminal em cinco a 10 anos10. Após transplante renal, a NM pode recorrer em cerca de 40% dos casos, reduzindo a sobrevida do enxerto11.
A escassez de marcadores diagnósticos e prognósticos, associados à evoluçao inconstante da NMI, dificultam a definiçao de quais pacientes com NMI devem receber tratamento específico, realizado com imunossupressores, podendo ter custo elevado e causar eventos adversos graves12. Sabe-se também que a NM pode apresentar-se como uma patologia renal isolada até anos antes de manifestaçoes sistêmicas de uma doença subjacente associada. Eventualmente, os achados clínicos e laboratoriais e histopatológicos renais nao sao suficientes para identificar a etiologia da doença13,14.
ENVOLVIMENTO DE IMUNOCOMPLEXOS NA FISIOPATOGENIA DA NM
A fisiopatogenia da NM nao está completamente elucidada, porém sabe-se que a proteinúria ocorre como consequência de depósitos imunes subepiteliais. A formaçao destes imunocomplexos pode ocorrer na circulaçao sistêmica, com posterior deposiçao glomerular, mecanismo principal de lesao renal na nefropatia membranosa secundária a lúpus eritematoso sistêmico. A formaçao de imunocomplexos pode ainda ocorrer in situ, como consequência da reaçao de anticorpos circulantes contra antígenos podocitários nativos ou exógenos, adquiridos principalmente durante a infância. Este é o mecanismo predominante na NMI2,15.
Os mecanismos de lesao renal na NMI vêm sendo estudados a partir do modelo experimental da nefrite de Heymann de 1952, responsável pela identificaçao da megalina podocitária como antígeno alvo para os anticorpos circulantes, levando à formaçao de imunocomplexos in situ16. A ausência da megalina em humanos, entretanto, levou à busca por novos antígenos. Os progressos dos estudos em humanos ocorreram quase 50 anos após, quando foram identificados novos antígenos podocitários em portadores de NMI.
Em 2002, Debiec et al. identificaram a endopeptidase neutra (NEP) como autoantígeno alvo em recém-natos portadores de NMI. Estes autores demonstraram a passagem de anticorpos anti-NEP através da placenta de maes deficientes em NEP, previamente sensibilizadas em outras gestaçoes com parceiros NEP-positivos17. Entretanto, por se tratar de uma rara apresentaçao da doença, e devido aos níveis séricos destes anticorpos serem semelhantes em adultos portadores de NMI comparados a indivíduos saudáveis18, a NEP isoladamente nao é capaz de explicar a fisiopatogenia da doença.
Em 2009, Beck et al. localizaram o receptor transmembrana tipo 1 de fosfolipase A2 do tipo M (PLA2R1), membro da família dos receptores de manose, nos podócitos de pacientes com NMI. Este antígeno é, até o momento, o que demonstra maior associaçao com a etiologia primária da NM, presente em 70% dos pacientes. Entretanto, cerca de 30% dos portadores de NMI permaneciam sem antígeno alvo identificado19. Dois anos após, a albumina sérica catiônica bovina foi identificada como possível antígeno podocitário exógeno, sendo adquirido e implantado na infância20. Alguns outros possíveis autoantígenos podocitários identificados foram aldolase redutase, superóxido dismutase 221, alfa-enolase22 e trombospondina23. É possível ainda que a NMI seja resultante da coexistência de mais de um alvo podocitário18.
ETIOPATOGENIA DA NM: UM POSSIVEL PAPEL ETIOPATOGENICO DA IGG4
O ponto em comum e intrigante dos antígenos podocitários descritos até o momento é que a maioria deles é alvo da IgG424. Esta imunoglobulina tem sua produçao regulada pelas células Th2 e está mais comumente associada a exposiçoes crônicas a antígenos25. É a subclasse menos abundante, em geral representando menos de 5% das IgGs, e possui características peculiares, como a heterobivalência. Esta propriedade resulta de uma sequência de aminoácidos (Cisteína-Prolina-Serina-Cisteína) mais susceptível a reduçao, causando a separaçao das cadeias pesadas do anticorpo e reassociaçao destas cadeias com outras moléculas de IgG4. Como consequência, formam-se moléculas bi-específicas, porém funcionalmente monovalentes, inibindo a formaçao de grandes imunocomplexos e reduzindo a capacidade de ativaçao da via clássica do complemento26.
Assim, algumas hipóteses buscando explicar a participaçao da IgG4 na fisiopatogenia da NMI têm sido propostas. Uma delas é a de que a IgG1, encontrada em fases mais precoces da NMI27, seja a responsável pela ativaçao da via clássica do complemento e, em seguida, da via alternativa e/ou das lectinas28. Neste caso, a IgG4 surgiria numa fase mais avançada da doença, como reflexo de uma reaçao crônica. Uma segunda hipótese está relacionada à natureza pró-inflamatória da IgG4 pobre em galactose terminal (IgG4-G0) e sua associaçao com o desenvolvimento de doenças autoimunes29. Este anticorpo serviria como epítopo para a ligaçao da proteína MBL, ativando a via das lectinas30.
Apesar das evidências do envolvimento local da IgG4 no glomérulo, estudos quanto a dosagem sérica das subclasses de IgG em NM sao escassos. De fato, Imai et al. (1997) em seu estudo nao encontraram diferença entre os níveis séricos das subclasses de IgG em 21 pacientes com NMI comparados a nove pacientes com NMS31. Entretanto, Kuroki et al. (2002) encontraram aumento significativo do percentual de IgG4 em relaçao a IgG sérica (%IgG4) nos pacientes portadores de NMI, comparados a um grupo controle e a portadores de NMS, os quais apresentaram aumento significativo do percentual de IgG132. Li et al. (2010) demonstraram ainda %IgG4 sérica maior em pacientes com NMI comparados a portadores de doença de lesoes mínimas e glomeruloesclerose segmentar e focal33.
Apesar da participaçao do sistema imune na NMI ser cada vez mais evidente na NMI, seu envolvimento nao foi completamente elucidado. Nenhum antígeno alvo identificado até o momento é capaz de explicar todos os casos da doença, e estudos a respeito da patogênese do complexo antígeno-anticorpo na NMI permanecem necessários. Sabe-se, também, que a presença destes imunocomplexos é capaz de ativar as vias do complemento, componente igualmente importante na fisiopatogenia da NMI.
ENVOLVIMENTO DA IGG4 E DAS VIAS DO COMPLEMENTO NA FISIOPATOGENIA DA NM
Desde os estudos iniciais de Heymann, é reconhecida a importância do sistema complemento para o desenvolvimento de NMI, a partir da constataçao de que ratos depletados de C3 nao apresentavam proteinúria. Posteriormente, estudos demonstraram a presença do complexo de ataque à membrana (C5b-9), via final do complemento34, bem como de componentes da via alternativa (C3) e clássica (C4d) em depósitos renais35,36. Entretanto, na NMI habitualmente nao há deposiçao de C1q, componente inicial da via clássica, e a via alternativa é incapaz de gerar C4d sozinha. Assim, a via das lectinas, cujo principal componente é a proteína MBL, parece ser uma potencial explicaçao para a presença de C4d na NMI, sem que haja ativaçao da via clássica1.
A MBL é uma proteína de fase aguda produzida primariamente no fígado, com papel importante na imunidade inata37 e no reconhecimento de autoantígenos, como células apoptoicas e necróticas38. Circula sob a forma de multímeros, cujas unidades básicas sao compostas por uma regiao N-terminal rica em cisteína, uma regiao de colágeno e um domínio de reconhecimento de carboidrato, com alta afinidade por moléculas com sacarídeos, como glicose, fucose, ma-nose, N-acetil-D-glucosamina, N-acetil-manosamina, mas nao galactose, presentes em diversos patógenos39. Esses multímeros sao estruturalmente semelhantes à molécula do C1q, sendo capazes de ativar a via clássica do complemento sem necessitar da ligaçao com anticorpos40.
Apesar de nao ser uma proteína investigada rotineiramente em glomerulopatias, existem evidências do envolvimento da MBL na NM. Em 1999, Lhotta, Wurzner e Konig, descreveram depósitos glomerulares desta proteína em 10 de 15 pacientes portadores de NMI, mas também em portadores de nefrite lúpica41. Posteriormente, Segawa et al. (2010) demonstraram depósitos mesangiais de MBL em oito de 10 pacientes com NMI42. A possível relaçao entre a NMI e a MBL nao está bem esclarecida. Devido à atraçao da MBL por carboidratos ligados a IgG4-G030 e a degalactosilaçao da IgG ocorrer com o envelhecimento43, aventa-se a possibilidade de que a produçao de anticorpos IgG4-G0 contra o PLA2R1 possa ser uma explicaçao para o predomínio da NMI em pacientes mais idosos1.
CONSIDERAÇOES FINAIS
Muito se aprendeu a respeito da fisiopatogenia da NMI desde o modelo experimental de Heymann, que descobriu a base molecular e os conceitos da deposiçao de complexos imunes e injúria glomerular da doença. É notável, entretanto, que décadas após o trabalho inicial, ainda nao há um modelo fisiopatogênico capaz de explicar a doença em todos os pacientes. Em conclusao, a NMI parece ser uma doença multifatorial, na qual mutaçoes genéticas levam a alteraçoes estruturais nas proteínas de células podocitárias que passam a servir como alvo para anticorpos circulantes do tipo IgG4, resultando na formaçao in situ de depósitos glomerulares imunes que ativam as vias do complemento (Figura 1).
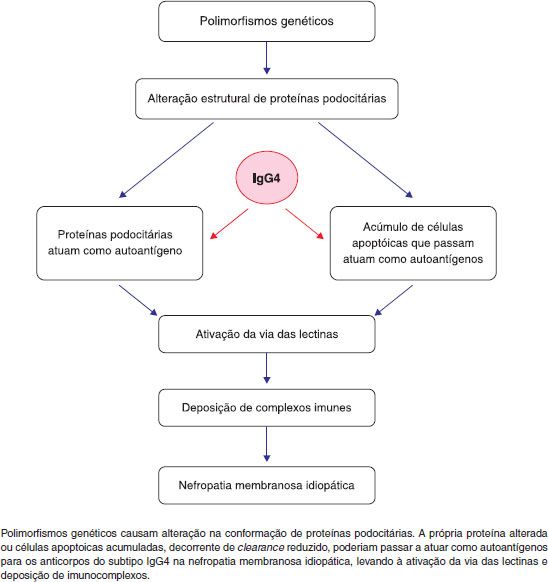
Figura 1 Modelo hipotético dos mecanismos etiopatogênicos da nefropatia membranosa idiopática
REFERENCIAS
1. Salant DJ. Genetic variants in membranous nephropathy: perhaps a perfect storm rather than a straightforward conformeropathy? J Am Soc Nephrol. 2013;24:525-8.
2. Glassock RJ. The pathogenesis of idiopathic membranous nephropathy: a 50-year odyssey. Am J Kidney Dis. 2010;56:157-67.
3. Diz MCE, Scherer P, Kirsztajn GM. Clinical-Epidemiological Profile of primary Membranous Glomerulopathy in Brazilian Patients (71 cases). J Bras Nefrol. 2007; 29:71-9.
4. Zeng CH, Chen HM, Wang RS, Chen Y, Zhang SH, Liu L, et al. Etiology and clinical characteristics of membranous nephropathy in Chinese patients. Am J Kidney Dis. 2008;52:691-8.
5. Maisonneuve P, Agodoa L, Gellert R, Stewart JH, Buccianti G, Lowenfels AB, et al. Distribution of primary renal diseases leading to end-stage renal failure in the United States, Europe, and Australia/New Zealand: results from an international comparative study. Am J Kidney Dis. 2000;35:157-65.
6. Malafronte P, Mastroianni-Kirsztajn G, Betônico GN, Romao JE Jr, Alves MA, Carvalho MF, et al. Paulista Registry of glomerulonephritis: 5-year data report. Nephrol Dial Transplant. 2006;21:3098-105.
7. Ronco P, Debiec H. Pathophysiological advances in membranous nephropathy: time for a shift in patient's care. Lancet. 2015 16;385:1983-92.
8. van den Brand JA, van Dijk PR, Hofstra JM, Wetzels JF. Long-term outcomes in idiopathic membranous nephropathy using a restrictive treatment strategy. J Am Soc Nephrol. 2014;25:150-8.
9. Polanco N, Gutiérrez E, Covarsí A, Ariza F, Carreño A, Vigil A, et al. Spontaneous remission of nephrotic syndrome in idiopathic membranous nephropathy J Am Soc Nephrol. 2010;21:697-704.
10. Glassock RJ. Diagnosis and natural course of membranous nephropathy. Semin Nephrol. 2003;23:324-32.
11. Sprangers B, Lefkowitz GI, Cohen SD, Stokes MB, Valeri A, Appel GB, et al. Beneficial effect of rituximab in the treatment of recurrent idiopathic membranous nephropathy after kidney transplantation. Clin J Am Soc Nephrol. 2010;5:790-7.
12. Hofstra JM, Fervenza FC, Wetzels JF. Treatment of idiopathic membranous nephropathy. Nat Rev Nephrol. 2013;9:443-58.
13. Sam R, Joshi A, James S, Jen KY, Amani F, Hart P, et al. Lupus-like membranous nephropathy: Is it lupus or not? Clin Exp Nephrol. 2015;19:395-402.
14. Murtas C, Ghiggeri GM. Membranous glomerulonephritis: histological and serological features to differentiate cancer-related and non-related forms. J Nephrol. 2016;29:469-78.
15. Ronco P, Debiec H. Pathogenesis of membranous nephropathy: recent advances and future challenges. Nat Rev Nephrol. 2012;8:203-13.
16. Heymann W, Lund HZ, Hackel DB. The nephrotic syndrome in the rats; with special reference to the progression of the glomerular lesion and to the use of nephrotoxic sera obtained from ducks. J Lab Clin Med. 1952;39:218-24.
17. Debiec H, Guigonis V, Mougenot B, Decobert F, Haymann JP, Bensman A, et al. Antenatal membranous glomerulonephritis due to anti-neutral endopeptidase antibodies. N Engl J Med. 2002;346:2053-60.
18. Murtas C, Bruschi M, Candiano G, Moroni G, Magistroni R, Magnano A, et al. Coexistence of different circulating anti-podocyte antibodies in membranous nephropathy. Clin J Am Soc Nephrol. 2012;7:1394-400.
19. Beck LH Jr, Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al. M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med. 2009;361:11-21.
20. Debiec H, Lefeu F, Kemper MJ, Niaudet P, Deschênes G, Remuzzi G, et al. Early-childhood membranous nephropathy due to cationic bovine serum albumin. N Engl J Med. 2011;364:2101-10.
21. Prunotto M, Carnevali ML, Candiano G, Murtas C, Bruschi M, Corradini E, et al. Autoimmunity in membranous nephropathy targets aldose reductase and SOD2. J Am Soc Nephrol. 2010;21:507-19.
22. Bruschi M, Carnevali ML, Murtas C, Candiano G, Petretto A, Prunotto M, et al. Direct characterization of target podocyte antigens and autoantibodies in human membranous glomerulonephritis: alfa-enolase and borderline antigens. J Proteomics. 2011;74:2008-17.
23. Tomas NM, Beck LH Jr, Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, Dolla G, Hoxha E, et al. Thrombospondin type-1 domaincontaining 7A in idiopathic membranous nephropathy. N Engl J Med. 2014;371:2277-87.
24. Stone JH. IgG4: a tantalizing link between causes of membranous glomerulonephritis and systemic disease. Kidney Int. 2013;83:348-50.
25. Nirula A, Glaser SM, Kalled SL, Taylor FR. What is IgG4? A review of the biology of a unique immunoglobulin subtype. Curr Opin Rheumatol. 2011;23:119-24.
26. Aalberse RC, Schuurman J. IgG4 breaking the rules. Immunology. 2002;105:9-19.
27. Huang CC, Lehman A, Albawardi A, Satoskar A, Brodsky S, Nadasdy G, et al. IgG subclass staining in renal biopsies with membranous glomerulonephritis indicates subclass switch during disease progression. Mod Pathol. 2013;26:799-805.
28. Thurman JM, Holers VM. The central role of the alternative complement pathway in human disease. J Immunol. 2006;176:1305-10.
29. Maverakis E, Kim K , Shimoda M , Gershwin ME, Patel F, Wilken R, et al. Glycans in the immune system and The Altered Glycan Theory of Autoimmunity: a critical review. J Autoimmun. 2015;57:1-13.
30. Malhotra R, Wormald MR, Rudd PM, Fischer PB, Dwek RA, Sim RB. Glycosylation changes of IgG associated with rheumatoid arthritis can activate complement via the mannose-binding protein. Nat Med. 1995;1:237-43.
31. Imai H, Hamai K, Komatsuda A, Ohtani H, Miura AB. IgG subclasses in patients with membranoproliferative glomerulonephritis, membranous nephropathy, and lupus nephritis. Kidney Int. 1997;51:270-6.
32. Kuroki A, Shibata T, Honda H, Totsuka D, Kobayashi K, Sugisaki T. Glomerular and serum IgG subclasses in diffuse proliferative lupus nephritis, membranous lupus nephritis, and idiopathic membranous nephropathy. Intern Med. 2002;41:936-42.
33. Li J, Qu Z, Zhang YM, Yu F, Huang J, Yang R, et al. Clinical significance of detection of plasma and urine IgG4 in idiopathic membranous nephropathy. Beijing Da Xue Xue Bao. 2010;42:671-4.
34. Cunningham PN, Quigg RJ. Contrasting roles of complement activation and its regulation in membranous nephropathy. J Am Soc Nephrol. 2005;16:1214-22.
35. Espinosa-Hernández M, Ortega-Salas R, López-Andreu M, Gómez- Carrasco JM, Pérez-Sáez MJ, Pérez-Seoane C, et al. C4d as a diagnostic tool in membranous nephropathy. Nefrologia. 2012;32:295-9.
36. Val-Bernal JF, Garijo MF, Val D, Rodrigo E, Arias M. C4d immunohistochemical staining is a sensitive method to confirm immunoreactant deposition in formalin-fixed paraffin-embedded tissue in membranous glomerulonephritis. Histol Histopathol. 2011;26:1391-7.
37. Bouwman LH, Roep BO, Roos A. Mannose-binding lectin: clinical implications for infection, transplantation, and autoimmunity. Hum Immunol. 2006;67:247-56.
38. Nauta AJ, Raaschou-Jensen N, Roos A, Daha MR, Madsen HO, Borrias-Essers MC, et al. Mannose-binding lectin engagement with late apoptotic and necrotic cells. Eur J Immunol. 2003;33:2853-63.
39. Turner MW. The role of mannose-binding lectin in health and disease. Mol Immunol. 2003;40:423-9.
40. Worthley DL, Bardy PG, Mullighan CG. Mannose-binding lectin: biology and clinical implications. Intern Med J. 2005;35:548-55.
41. Lhotta K, Würzner R, König P. Glomerular deposition of mannosebinding lectin in human glomerulonephritis. Nephrol Dial Transplant. 1999;14:881-6.
42. Segawa Y, Hisano S, Matsushita M, Fujita T, Hirose S, Takeshita M, et al. IgG subclasses and complement pathway in segmental and global membranous nephropathy. Pediatr Nephrol. 2010;25:1091-9.
43. Pucić M, Knezević A, Vidic J, Adamczyk B, Novokmet M, Polasek O, et al. High throughput isolation and glycosylation analysis of IgG-variability and heritability of the IgG glycome in three isolated human populations. Mol Cell Proteomics. 2011;10:M111.010090.
44. Klouda PT, Manos J, Acheson EJ, Dyer PA, Goldby FS, Harris R, et al. Strong association between idiopathic membranous nephropathy and HLA-DRW3. Lancet. 1979;2:770-1.
45. Vaughan RW, Demaine AG, Welsh KI. A DQA1 allele is strongly associated with idiopathic membranous nephropathy. Tissue Antigens. 1989;34:261-9.
46. Liu YH, Chen CH, Chen SY, Lin YJ, Liao WL, Tsai CH, et al. Association of phospholipase A2 receptor 1 polymorphisms with idiopathic membranous nephropathy in Chinese patients in Taiwan. J Biomed Sci. 2010;17:81.
47. Stanescu HC, Arcos-Burgos M, Medlar A, Bockenhauer D, Kottgen A, Dragomirescu L, et al. Risk HLA-DQA1 and PLA(2)R1 alleles in idiopathic membranous nephropathy. N Engl J Med. 2011;364:616-26.
48. Coenen MJ, Hofstra JM, Debiec H, Stanescu HC, Medlar AJ, Stengel B, et al. Phospholipase A2 receptor (PLA2R1) sequence variants in idiopathic membranous nephropathy. J Am Soc Nephrol. 2013;24:677-83.
49. Bally S, Debiec H, Ponard D, Dijoud F, Rendu J, Fauré J, et al. Phospholipase A2 Receptor-Related Membranous Nephropathy and Mannan-Binding Lectin Deficiency. J Am Soc Nephrol. 2016;27(12):3539-44.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888