Número Atual: Março-Abril 2015 - Volume 3 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

RELATO DE CASO
Imunodeficiência comum variável: dificuldades no diagnóstico
Common variable immunodeficiency: diagnostic constraints
Karin Milleni Araujo, MD; Licio Augusto Velloso, MD, PhD; Eli Mansour, MD, PhD
DOI: 10.5935/2318-5015.20150013
Departamento de Clínica Médica, Faculdade de Ciências Médicas, Universidade Estadual de Campinas, UNICAMP, Campinas, Sao Paulo, Brasil
Endereço para correspondência:
Karin Milleni Araujo
E-mail: karin_milleni@hotmail.com
Submetido em: 03/10/2015.
Aceito em: 05/07/2016.
Nao foram declarados conflitos de interesse associados à publicaçao deste artigo.
RESUMO
A imunodeficiência comum variável (IDCV) é a imunodeficiência primária sintomática mais comum, e representa um conjunto heterogêneo de distúrbios que resultam principalmente da deficiência de anticorpos, levando a infecçoes recorrentes. A variabilidade na expressao clínica e o desconhecimento da doença contribuem para o retardo no diagnóstico, aumentando a morbidade e a mortalidade. Neste artigo apresentamos o caso clínico de um jovem diagnosticado com IDCV, ressaltando as dificuldades para se estabelecer o diagnóstico frente aos múltiplos achados clínicos e laboratoriais durante o processo de investigaçao. Tal fato levou a hospitalizaçao prolongada, com grande número de complicaçoes graves e elevado custo.
Descritores: Imunodeficiência comum variável, manifestaçoes clínicas, critérios diagnósticos, diagnóstico tardio.
INTRODUÇAO
A imunodeficiência comum variável (IDCV) é uma imunodeficiência primária caracterizada pela diminuiçao de imunoglobulinas e pode apresentar ou nao diminuiçao de linfócitos B e T1-3. Tem sido considerada uma afecçao rara1,2, entretanto, acredita-se que sua incidência seja subestimada principalmente devido à falta de diagnóstico correto1,3.
Nesse contexto, por representar um conjunto heterogêneo de manifestaçoes clínicas, a IDCV tem sido identificada tardiamente mesmo por profissionais experientes1-3. Tais dificuldades diagnósticas acarretam atraso no início do tratamento adequado e, portanto, aumento nas taxas de hospitalizaçao, morbidade e mortalidade1-3. Logo, é importante que a IDCV seja conhecida pela classe médica, para que entao faça parte do raciocínio diagnóstico e abordagem diagnóstica específica seja prontamente estabelecida, evitando-se o desenvolvimento de complicaçoes graves, muitas vezes, irreversíveis1,2.
O objetivo deste artigo é relatar um paciente diagnosticado com IDCV ressaltando as dificuldades para se estabelecer o diagnóstico frente aos múltiplos achados clínicos e laboratoriais durante o processo de investigaçao, o que levou a hospitalizaçao prolongada e elevado custo.
DESCRIÇAO DO CASO
Paciente do sexo masculino, 26 anos, procurou atendimento em nosso Serviço de Urgência/Emergência devido a quadro febril intermitente associado a sintomas predominantemente respiratórios que iniciaram há um mês. Durante esse período recebeu atendimento médico em outros serviços e permaneceu internado por 11 dias sob ventilaçao mecânica por insuficiência respiratória aguda, sem o estabelecimento de diagnóstico. Referia piora dos sintomas há uma semana, com febre diária predominantemente noturna, tosse com expectoraçao, e otalgia com otorreia à direita.
Ao exame físico, apresentava-se febril e taquicárdico. Otoscopia direita revelou secreçao serosa em grande quantidade associada a edema de conduto auditivo com membrana timpânica íntegra; sem sinais clínicos sugestivos de mastoidite. Ritmo cardíaco regular em dois tempos, com bulhas normofonéticas e sopro sistólico em foco aórtico. Fígado palpável a três centímetros do rebordo costal direito e baço percutível. Foi identificado um linfonodo axilar à esquerda de dois centímetros, indolor, fibroelástico e móvel. O restante do exame físico nao apresentava alteraçoes. Negou antecedentes familiares significativos.
Nesse momento, foram aventadas hipóteses de possíveis focos infecciosos tais como abscesso, endocardite, tuberculose ou meningite. O paciente foi entao internado para investigaçao adicional. A investigaçao laboratorial revelou: hemoglobina 9,8 g/dL, hematócrito 31%, volume corpuscular médio 86,4 fl e reticulócitos 14,3%; leucócitos 7.340/mm3, segmentados 83%, bastonetes 6%, linfócitos 1,2%, monócitos 0%, eosinófilos 1,2% e basófilos 0%; esfregaço sanguíneo com discreta policromatofilia; e proteína C-reativa 13,9 mg/dL e velocidade de hemossedimentaçao 49 mm na primeira hora. Culturas de líquidos biológicos (sangue, urina, escarro e liquor) sem crescimento de micro-organismos. Sorologias para os vírus das hepatites A, B e C, e para o vírus da imunodeficiência humana foram negativas, além de anti-HBs e sorologias (IgM e IgG) para citomegalovírus, vírus Epstein Barr e toxoplasmose, evidenciando uma resposta pobre ou ausente às imunizaçoes. Ecocardiografias cardíacas transtorácica e transesofágica foram normais. Tomografias computadorizadas de crânio-face, tórax e abdome revelaram sinusite maxilar bilateral, algumas áreas de bronquiectasia discretas e hepatoesplenomegalia moderada.
Durante a internaçao, o paciente evoluiu com febre alta diária associada a episódios de bacteremia. Dez dias após a admissao, apresentou-se com síndrome da angústia respiratória aguda e choque séptico com disfunçao de múltiplos órgaos. Foi entao transferido para unidade de terapia intensiva, ainda sem diagnóstico definido. Iniciou-se antibioticoterapia empírica de largo espectro, evoluindo com melhora da curva térmica e dos parâmetros hemodinâmicos e ventilatórios.
Nesse ponto da investigaçao foram levantadas novas hipóteses diagnósticas, tais como neoplasia hematológica, doenças reumatológicas e doenças autoimunes. As equipes de hematologia e reumatologia avaliaram o paciente. Biópsia de medula óssea descartou afecçao hematológica. Biópsia de mucosa nasal evidenciou colonizaçao por Candida albicans, Pseudomonas aeruginosa e Acinetobacter baumannii sem outras alteraçoes. Autoanticorpos negativos (fator antinuclear, fator reumatoide, anticorpo antiantígeno nuclear extraível e anticorpos anticitoplasma de neutrófilo) afastaram doenças reumatológicas.
O paciente manteve disfunçoes orgânicas em progressao, apesar de cobertura antibiótica. Após extensa busca diagnóstica sem soluçao do caso, foi entao aventada hipótese de imunodeficiência primária. Foram solicitadas dosagem de imunoglobulinas por nefelometria: imunoglobulina G 401 mg/dL, imunoglobulina A < 6,59 mg/dL e imunoglobulina M 56,8 mg/dL (Referências para a idade [Laboratório HC UNICAMP]: IgG 830-2040 mg/dL; IgA 80-476 mg/dL; IgM 57-212 mg/dL). Nesse momento, a equipe de imunologia, baseada na combinaçao da hipogama-globulinemia e os demais achados clínicos, realizou o diagnóstico de imunodeficiência humoral primária, em especial a IDCV. Imunofenotipagem linfocitária realizada por meio de citometria de fluxo apresentou: linfócitos 5390/mm3, CD3 76,6% (513 cels/mm3), CD19 2,2% (14 cels/mm3), CD3/CD4 26,1% (174 cels/mm3), CD3/CD8 45,1% (302 cels/mm3), CD16+CD56 13,4% (89 cels/mm3), CD3/CD16+CD56 1,7% (11 cels/mm3) e relaçao CD4/CD8 invertida (0,57). Assim, outras imunodeficiências primárias foram excluídas e, portanto, confirmando-se a hipótese de IDCV. Iniciou-se entao reposiçao de imunoglobulina humana intravenosa (500 mg/kg) com rápida recuperaçao das disfunçoes secundárias ao quadro séptico, sendo possível alta 30 dias após admissao hospitalar. Ultima dosagem de imunoglobulinas realizada com dois anos de acompanhamento apresenta IgG 647 mg/dL, IgA < 6,0 mg/dL, IgM < 16,8 mg/dL e IgE < 4,45 UI/mL. O paciente continua em seguimento ambulatorial com a equipe de imunologia e encontra-se em tratamento com reposiçao periódica de imunoglobulina humana (500 mg/kg) desde o diagnóstico, mantendo estabilidade clínica e sem novas infecçoes graves.
DISCUSSAO
A IDCV é a imunodeficiência primária mais prevalente e sintomática em crianças e adultos, sem diferenças entre os sexos1-2. A incidência estimada é de 1 em cada 10.000-100.000 sujeitos1. A idade do início dos sintomas é variável, apresentando dois picos principais de incidência - na infância e entre a segunda e a terceira década de vida (mais comum)4,5. Porém o diagnóstico geralmente tem sido corretamente efetuado, em média, 6-7 anos após o início dos sintomas, principalmente devido à variabilidade de apresentaçao clínica2,6. Ademais, a falta de suspeita por desconhecimento da classe médica também tem sido reportada como um fator adicional importante no atraso diagnóstico1,2.
Clinicamente, a IDCV é caracterizada por um amplo espectro de manifestaçoes1-3. A apresentaçao clínica típica da IDCV é de infecçoes bacterianas recorrentes do trato respiratório, como reportado em nosso paciente, mas outras infecçoes podem também ocorrer1-3. As infecçoes sinopulmonares (por exemplo, pneumonias, bronquites, sinusites, otites e conjuntivites) e gastrointestinais (principalmente diarreia por Giardia lamblia) secundárias, em sua maioria, a agentes bacterianos, sao as infecçoes mais comumente identificadas e vivenciadas por esses pacientes1,2. Tem sido relatado que 90% dos pacientes com IDCV sofre um ou mais episódios de infecçoes do trato respiratório inferior antes do diagnóstico. Os organismos mais comumente isolados sao Streptococcus pneumoniae e Haemophilus influenzae1. As doenças pulmonares crônicas sao a maior causa de hospitalizaçoes recorrentes, contribuindo com o elevado índice de morbidade e mortalidade1,7. Aproximadamente um terço dos pacientes já apresenta algum grau de doença pulmonar crônica ao diagnóstico de IDCV1,7.
Ademais, a IDCV também apresenta aspectos da alteraçao no sistema imunológico com complicaçoes nao infecciosas, incluindo na autoimunidade (mais tipicamente citopenias autoimunes - trombocitopenia autoimune e anemia hemolítica autoimune, e mais raramente a neutropenia autoimune), doença gastrointestinal nao infecciosa, doenças granulomatosas e proliferaçoes linfoides, que estao associadas a um pior prognóstico1,2.
Nossa hipótese diagnóstica inicial para esse caso foi de um processo neoplásico, descartada após investigaçao específica; uma alta susceptibilidade ao câncer também tem sido reportada em pacientes com ICVD1,2. As doenças malignas apresentam incidência global elevada na IDCV, e certos tipos de câncer sao significativamente mais comuns, particularmente carcinoma gástrico e linfomas nao-Hodgkin, que têm uma incidência 7-16 e 12-18 vezes maior, respectivamente, dependendo do estudo1,7. As razoes para o elevado risco de malignidade na IDCV sao multifatoriais, e podem estar potencialmente ligadas a interaçoes complexas entre a estimulaçao antigênica crônica pelas infecçoes, a aquisiçao de anormalidades genéticas e as alteraçoes no sistema imunológico1.
O exame físico dos pacientes com IDCV pode ser normal ou apresentar apenas achados de doenças crônicas, tais como retardo no crescimento ou perda de peso1,2,8. Outros achados comuns incluem descarga nasal ou congestao significativa secundária a sinusite crônica, além de linfadenopatia, esplenomegalia, artrite, e alteraçoes dermatológicas associadas a doenças autoimunes8. Nosso paciente apresentava inúmeros desses sinais e sintomas, caracterizados tanto pela história e exame clínico, como por exames complementares.
Pacientes com IDCV geralmente nao apresentam anormalidades nos exames de rotina. De fato, inúmeros exames como, por exemplo, marcadores sorológicos para triagem de infecçoes virais e marcadores reumatológicos, nao auxiliaram efetivamente na elucidaçao diagnóstica, como descrito previamente1,2,6. Ocasionalmente pode ser observada reduçao das globulinas séricas e/ou no nível total de proteínas e linfopenia discreta9,10.
A IDCV é caracterizada por uma deficiência primária de anticorpos (hipogamaglobulinemia) de pelo menos dois isotipos de imunoglobulinas. Imunoglobulinas séricas sao marcadamente anormais em pacientes com IDCV. IgG sérica apresenta-se abaixo do limite normal1,3; particularmente em adultos um limite inferior a 450 mg/dL foi recentemente proposto1. Adicionalmente, IgA e/ou IgM também devem apresentar valores abaixo da normalidade1,3. Metade dos pacientes pode nao apresentar níveis detectáveis de imunoglobulinas1.3. Os níveis de IgG devem estar reduzidos, no mínimo, em 2 ocasioes com mais de 3 semanas de intervalo1. Além de IgG abaixo da normalidade, o paciente reportado aqui também apresentava IgA, IgM e IgE com valores abaixo da normalidade ou com níveis indetectáveis. A maioria dos pacientes tem números normais de linfócitos T e B; entretanto, podem apresentar alguma reduçao nas células B de memória1,2. A dosagem de anticorpos específicos nao foi realizada no presente caso por indisponibilidade no serviço. Na literatura, tais análises nao têm sido consideradas obrigatórias para o estabelecimento diagnóstico1,11,12.
Como descrito previamente1,3, o caso clínico apresentado acima ilustra a constelaçao de sintomas comuns encontrados em pacientes com IDCV, assim como de inúmeros encaminhamentos para especialidades diversas, contribuindo para o atraso no diagnóstico comumente visto nesta doença. Como a apresentaçao da IDCV é extremamente heterogênea, os pacientes geralmente procuram o primeiro atendimento em diferentes especialistas, por exemplo, otorrinolaringologistas, pneumologistas, gastroenterologistas e reumatologistas2,6. Entretanto, como os defeitos imunes primários têm sido mais comumente relacionados às doenças pediátricas, a maioria dos médicos (principalmente nao pediatras) parece nao ser familiarizada com a IDCV e, portanto, existem atrasos diagnósticos2, como o reportado no presente caso.
Nesse contexto, inúmeras campanhas informativas como, por exemplo, "J-Project", "Is it PID?", "FIND ID" e "The 10 warning signs" vêm sendo lançadas em diferentes locais ( Europa Central, Reino Unido, Alemanha e Estados Unidos, respectivamente) com o intuito de aprimorar o conhecimento, reduzir o diagnóstico tardio, melhorar o tratamento e prevenir as complicaçoes secundárias à IDCV1,6. No Brasil, o Grupo Brasileiro de Imunodeficiências (BRAGID) tem sido o responsável por iniciativas semelhantes, em parceria com a Fundaçao Jeffrey Modell e a Associaçao Brasileira de Alergia e Imunologia13.
Para suspeita clínica de novos casos de imunodeficiências primárias no adulto, sao utilizados os seguintes critérios com base nos sinais de alerta da European Society for Immunodeficiencies (ESID) - Primary Immunodeficiency Diseases (Tabelas 1 e 2)12, adaptados para o Brasil13: 1) duas ou mais novas otites no período de um ano; 2) duas ou mais novas sinusites no período de um ano, na ausência de alergia; 3) uma pneumonia por ano por mais que um ano; 4) diarreia crônica com perda de peso; 5) infecçoes virais de repetiçao (resfriados, herpes, verrugas, condiloma); 6) uso de antibiótico intravenoso de repetiçao para o tratamento de infecçoes; 7) abscessos profundos de repetiçao na pele ou em órgaos internos; 8) monilíase persistente ou infecçao fúngica na pele ou qualquer outro lugar; 9) infecçao por Micobacterium tuberculosis ou atípica; 10) história familiar de imunodeficiência.


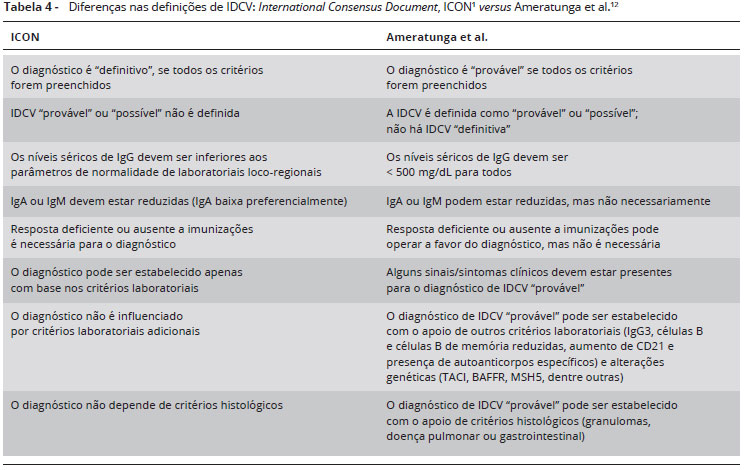
Além disso, existe uma série de novos critérios diagnósticos propostos para auxiliar na melhor identificaçao e classificaçao dos pacientes (Tabelas 1, 2, 3 e 4)1,12,14. Nao se pode deixar de ressaltar a importância de nao se limitar aos sinais de alerta para imunodeficiências primárias, e muito menos ao número de infecçoes, pois neste caso a sensibilidade é de 56%, logo, é importante suspeitar de imunodeficiência primária em qualquer infecçao cujo comportamento foge do usual1,14.

Semelhante ao que foi descrito em nosso caso, o diagnóstico da IDVC é estabelecido na presença de reduçao significativa na concentraçao sérica de IgG combinada com baixos níveis de IgA e/ou IgM e resposta pobre ou ausente às imunizaçoes, tendo sido excluídas outras causas de hipogamaglobulinemia1,14, sendo por isso vista como um diagnóstico de exclusao1. Portanto, em todos os pacientes com história e exame físico sugestivos de um possível diagnóstico de IDCV, deve se realizar uma dosagem de imunoglobulinas séricas e realizar o referenciamento do paciente ao imunologista o quanto antes, para que sejam realizados testes adicionais, excluam-se outras causas de hipogamaglobulinemia e a terapia mais apropriada possa ser instituída2,11.
É fundamental o diagnóstico precoce para que tratamento adequado seja iniciado o quanto antes, reduzindo o número de infecçoes secundárias e hospitalizaçoes e, consequentemente, a morbimortalidade global1,8,10,15. O tratamento da IDCV é baseado na reposiçao periódica de imunoglobulina intravenosa (IGIV) ou imunoglobulina subcutânea (IGSC)1,12,15. De acordo com classificaçao proposta por Ameratunga et al.12,14 (Tabela 3), os pacientes suspeitos para IDCV deveriam ser divididos em casos "possíveis" ou "prováveis". Os pacientes precisam apresentar todos os critérios maiores na categoria "A" para que seja considerada a hipótese de IDCV12,14; a categoria "B" confirma a presença de sintomas indicando falência do sistema imune. Para diagnosticar IDCV "provável", os pacientes também devem apresentar evidências laboratoriais que comprovem a disfunçao do sistema imune (categoria "C") ou lesoes histológicas características (categoria "D"). Pacientes com hipogamaglobulinemia leve (IgG > 500 mg/dL) sao denominados com hipogamaglobulinemia de significado incerto (HGSI). Pacientes que preenchem os critérios da categoria "A", mas nao os outros sao denominados de IDCV "possível". A maioria dos pacientes com IDCV "provável" deve ser tratada com IGIV/IGSC. Alguns pacientes com IDCV "possível" que apresentam hipogamaglobulinemia poderao ser tratados com IGIV/IGSC, contudo a maioria dos pacientes com HGSI nao precisará de reposiçao (Figura 1)12.
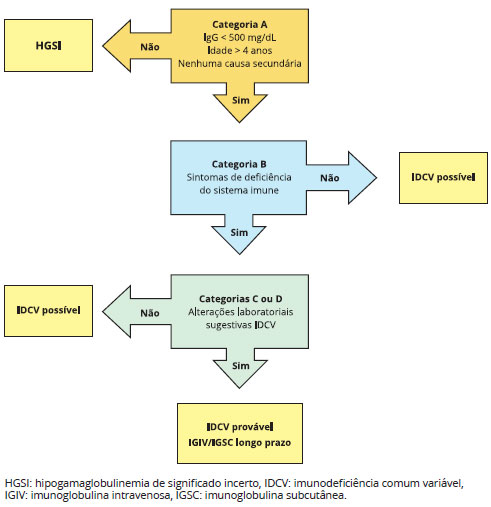
Figura 1 - Algoritmo terapêutico recentemente publicado por Ameratunga et al.12,14
Nesse contexto, o Consenso Internacional para IDCV (International Consensus Document, ICON) recentemente elaborado por Bonilla et al.1 nao classifica o diagnóstico de IDCV em "possível" ou "provável" (Tabela 4). Porém, é importante destacar que alguns pacientes com hipogamaglobulinemia e resposta pobre ou ausente a imunizaçoes possam nao preencher todos os critérios para o diagnóstico de IDCV, apresentando níveis séricos normais de IgA ou IgM, principalmente inicialmente1. Ademais, Bonilla et al.1 reportaram que na vigência de níveis muito baixos de IgG e clínica exuberante (muito sugestiva de IDCV), a reposiçao de imunoglobulina está indicada sem a necessidade de dosar anticorpos específicos.
A terapia de substituiçao com imunoglobulina representa a base do tratamento na IDCV1,12,15. É a intervençao médica mais efetiva na reduçao de infecçoes bacterianas, hospitalizaçoes e lesoes crônicas de órgaos1,4,15. A reposiçao de imunoglobulina também pode melhorar substancialmente a qualidade de vida e longevidade dos pacientes com IDCV12,14. Nao há nenhum consenso quanto à antibioticoprofilaxia, mas a vacina anual contra influenza (inativada) tem sido recomendada6. Embora os pacientes com IDCV apresentem risco elevado para o desenvolvimento de câncer, nao existe nenhum protocolo específico para triagem1,14. Nosso paciente recebeu imunoglobulina com melhora progressiva do quadro séptico. Continua em seguimento em nosso serviço, e há 36 meses nao apresentou nenhuma complicaçao ou recidiva clinicamente detectável.
CONCLUSAO
Reportamos o diagnóstico de IDCV em um adulto jovem. O diagnóstico precoce da IDCV é extremamente importante, pois o tratamento retardado ou inadequado pode levar ao aumento de complicaçoes irreversíveis, piora na qualidade de vida e, por fim, à mortalidade precoce. Logo, é importante que os médicos estejam cientes das manifestaçoes clínicas para que o tempo entre o estabelecimento do diagnóstico e o início da terapia seja reduzido. Reforçamos a necessidade de continuar o trabalho de divulgar as imunodeficiências primárias para os profissionais da saúde (especialmente médicos), assim como alertá-los de que várias imunodeficiências primárias podem ter início em adultos.
REFERENCIAS
1. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. The many faces of common variable immunodeficiency. j Allergy Clin Immunol Pract. 2016;4:38-59.
2. Cunningham-Rundles C, Maglione PJ. Common variable immunodeficiency. J Allergy Clin Immunol. 2012;129:1425-6.
3. Resnick ES, Cunningham-Rundles C. The many faces of the clinical picture of common variable immune deficiency. Curr Opin Allergy Clin Immunol. 2012;12:595-601.
4. Abolhassani H, Amirkashani D, Parvaneh N, Mohammadinejad P, Gharib B, Shahinpour S, et al. Autoimmune phenotype in patients with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2013;23:323-9.
5. Lourdes LS, Daily KC. Common variable immunodeficiency syndrome in an adult. Lancet. 2014;383:926.
6. Yesillik S, Musabak U, Sener O, Baysan A, Ucar E, Demirel F, et al. The diagnosis of common variable immunodeficiency in adults should not be missed: a delayed diagnosis can be devastating. Allergol Immunopathol. 2014;42:620-2.
7. Maarschalk-Ellerbroek LJ, Hoepelman AI, Van Montfrans JM, Ellerbroek PM. The spectrum of disease manifestations in patients with common variable immunodeficiency disorders and partial antibody deficiency in a university hospital. J Clin Immunol. 2012;32:907-21.
8. Ramírez-Vargas N, Arablin-Oropeza SE, Mojica-Martínez D, Yamazaki-Nakashimada MA, de la Luz García-Cruz M, Terán-Juárez LM et al. Clinical and immunological features of common variable immunodeficiency in Mexican patients. Allergol Immunopathol. 2014;42:235-40.
9. Gathmann B, Mahlaoui N, Ceredih GL, Oksenhendler E, Warnatz K, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol. 2014;134:116-26.
10. Tam JS, Routes JM. Common variable immunodeficiency. Am J Rhinol Allergy. 2013;27:260-5.
11. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for common variable immune deficiency (CVID), which may assist with decisions to treat with intravenous or subcutaneous immunoglobulin. Clin Exp Immunol. 2013;174:203-11.
12. Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. 2014;5:415.
13. Grupo Brasileiro de Imunodeficiências. BRAGID. Os dez sinais de alerta para imunodeficiências primárias. Available from: http://www.imunopediatria.org.br/ [accessed 22.05.11].
14. Costa-Carvalho BT, Grumach AS, Franco JL, Espinosa-Rosales FJ, Leiva LE, King A, et al. Attending to Warning Signs of Primary Immunodeficiency Diseases Across the Range of Clinical Practice. J Clin Immunol. 2014;34:10-22.
15. Costa-Carvalho BT, Solé D, Condino-Neto A, Rosário Filho N. I Consenso Brasileiro sobre o uso de Imunoglobulina Humana em pacientes com Imunodeficiência Primárias. Sociedade Brasileira de Alergia e Imunopatologia. Rev Bras Alerg Imunol. 2010;33:104-16.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888