Número Atual: Abril-Junho 2022 - Volume 6 - Número 2
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Régis A. Campos
- Faradiba Sarquis Serpa
- Eli Mansour
- Maria Luiza Oliva Alonso
- Luisa Karla Arruda
- Marcelo Vivolo Aun
- Maine Luellah Demaret Bardou
- Ana Flávia Bernardes
- Fernanda Lugão Campinhos
- Herberto Jose Chong-Neto
- Rosemeire Navickas Constantino-Silva
- Jane da Silva
- Sérgio Duarte Dortas-Junior
- Mariana Paes Leme Ferriani
- Joanemile Pacheco de Figueiredo
- Pedro Giavina-Bianchi
- Lais Souza Gomes
- Ekaterini Goudouris
- Anete Sevciovic Grumach
- Marina Teixeira Henriques
- Antônio Abilio Motta
- Therezinha Ribeiro Moyses
- Fernanda Leonel Nunes
- Jorge A. Pinto
- Nelson Augusto Rosario-Filho
- Norma de Paula M. Rubini
- Almerinda Maria do Rêgo Silva
- Dirceu Solé
- Ana Julia Ribeiro Teixeira
- Eliana Toledo
- Camila Lopes Veronez
- Solange Oliveira Rodrigues Valle

ARTIGO ESPECIAL
Diretrizes brasileiras do angioedema hereditário 2022 - Parte 1: definição, classificação e diagnóstico
2022 Brazilian guidelines for hereditary angioedema - Part 1: definition, classification, and diagnosis
Régis A. Campos1; Faradiba Sarquis Serpa2; Eli Mansour3; Maria Luiza Oliva Alonso4; Luisa Karla Arruda5; Marcelo Vivolo Aun6,7; Maine Luellah Demaret Bardou8; Ana Flávia Bernardes3; Fernanda Lugão Campinhos2; Herberto Jose Chong-Neto9; Rosemeire Navickas Constantino-Silva10; Jane da Silva11; Sérgio Duarte Dortas-Junior4; Mariana Paes Leme Ferriani5; Joanemile Pacheco de Figueiredo12; Pedro Giavina-Bianchi6; Lais Souza Gomes6; Ekaterini Goudouris13; Anete Sevciovic Grumach8; Marina Teixeira Henriques8; Antônio Abilio Motta6; Therezinha Ribeiro Moyses2; Fernanda Leonel Nunes5; Jorge A. Pinto14; Nelson Augusto Rosario-Filho9; Norma de Paula M. Rubini15; Almerinda Maria do Rêgo Silva16; Dirceu Solé17; Ana Julia Ribeiro Teixeira6; Eliana Toledo18; Camila Lopes Veronez19; Solange Oliveira Rodrigues Valle4
DOI: 10.5935/2526-5393.20220019
1. Faculdade de Medicina da Bahia, Universidade Federal da Bahia, Departamento de Medicina Interna e Apoio Diagnóstico, Programa de Pós-graduação em Ciências da Saúde - Salvador, BA, Brasil
2. Escola Superior de Ciências da Santa Casa de Misericórdia de Vitória, Serviço de Referência em Asma, Alergia e Imunologia - Vitória, ES, Brasil
3. Faculdade de Ciências Médicas, Universidade Estadual de Campinas, Divisão de Alergia e Imunologia Clínica, Departamento de Clínica Médica - Campinas, SP, Brasil
4. Hospital Universitário Clementino Fraga Filho da Universidade Federal do Rio de Janeiro, Serviço de Imunologia - Rio de Janeiro, RJ, Brasil
5. Faculdade de Medicina de Ribeirão Preto - Universidade de São Paulo, Disciplina de Imunologia Clínica e Alergia, Departamento de Clínica Médica - Ribeirão Preto, SP, Brasil
6. Faculdade de Medicina da Universidade de São Paulo, Disciplina de Imunologia Clínica e Alergia - São Paulo, SP, Brasil
7. Faculdade Israelita de Ciências da Saúde Albert Einstein, Disciplina Agente Hospedeiro - São Paulo, SP, Brasil
8. Centro Universitário Faculdade de Medicina do ABC, Disciplina de Imunologia Clínica - Santo André, SP, Brasil
9. Universidade Federal do Paraná, Serviço de Alergia e Imunologia, Departamento de Pediatria - Curitiba, PR, Brasil
10. Centro Universitário Faculdade de Medicina do ABC, Laboratório de Imunologia Clínica - Santo André, SP, Brasil
11. Hospital Universitário Prof. Polydoro Ernani de São Thiago, Departamento de Clínica Médica da Universidade Federal de Santa Catarina - Florianópolis, SC, Brasil
12. Faculdade de Medicina da Bahia, Universidade Federal da Bahia, Departamento de Medicina Interna e Apoio Diagnóstico - Salvador, BA, Brasil
13. Faculdade de Medicina da Universidade Federal do Rio de Janeiro, Departamento de Pediatria - Rio de Janeiro, RJ, Brasil
14. Hospital das Clínicas da Faculdade de Medicina da Universidade Federal de Minas Gerais, Serviço de Imunologia Clínica - Belo Horizonte, MG, Brasil
15. Escola de Medicina e Cirurgia da Universidade Federal do Estado do Rio de Janeiro - UNIRIO, Disciplina de Alergia e Imunologia - Rio de Janeiro, RJ, Brasil
16. Universidade Federal de Pernambuco, Área Acadêmica de Pediatria, Centro de Ciências Médicas - Recife, PE, Brasil
17. Escola Paulista de Medicina - Universidade Federal de São Paulo, Disciplina de Alergia, Imunologia e Reumatologia - Departamento de Pediatria - São Paulo, SP, Brasil
18. Faculdade de Medicina de São José do Rio Preto, Serviço de Alergia e Imunologia Clínica do Departamento de Pediatria e Cirurgia Pediátrica - São José do Rio Preto, SP, Brasil
19. University of California, Divisão de Reumatologia, Alergia e Imunologia, Departamento de Medicina - San Diego, Califórnia, EUA
Endereço para correspondência:
Régis A. Campos
E-mail: regiscampos@ufba.br
Submetido em: 02/04/2022
Aceito em: 08/04/2022
Conflito de interesses Régis A. Campos, Faradiba S. Serpa, Maria Luisa O. Alonso, Pedro Giavina-Bianchi, Herberto José Chong-Neto, Eli Mansour, Eliana Toledo, Anete S. Grumach e Solange O. R. Valle receberam apoio financeiro e/ou honorários da Takeda e CSL Behring. Anete S. Grumach é bolsista de produtividade CNPq e também fez consultoria para a Catalyst. Receberam apoio financeiro e/ou honorários da Takeda os seguintes autores: Camila L. Veronez, Jane da Silva, Marcelo V. Aun, L. Karla Arruda. Os demais autores negam conflitos de interesse
RESUMO
O angioedema hereditário é uma doença autossômica dominante caracterizada por crises recorrentes de edema que acometem o tecido subcutâneo e o submucoso, com envolvimento de diversos órgãos. Os principais locais afetados são face, membros superiores e inferiores, as alças intestinais e as vias respiratórias superiores. Em decorrência da falta de conhecimento dessa condição por profissionais de saúde, ocorre atraso importante no seu diagnóstico, comprometendo a qualidade de vida dos indivíduos afetados. Além disso, o retardo no diagnóstico pode resultar em aumento da mortalidade por asfixia devido ao edema de laringe. A natureza errática das crises com variação do quadro clínico e gravidade dos sintomas entre diferentes pacientes, e no mesmo paciente ao longo da vida, se constitui em desafio no cuidado dos doentes que têm angioedema hereditário. O principal tipo de angioedema hereditário é resultante de mais de 700 variantes patogênicas do gene SERPING1 com deficiência funcional ou quantitativa da proteína inibidor de C1, porém nos últimos anos outras mutações foram descritas em seis outros genes. Ocorreram avanços importantes na fisiopatologia da doença e novas drogas para o tratamento do angioedema hereditário foram desenvolvidas. Nesse contexto, o Grupo de Estudos Brasileiro em Angioedema Hereditário (GEBRAEH) em conjunto com a Associação Brasileira de Alergia e Imunologia (ASBAI) atualizou as diretrizes brasileiras do angioedema hereditário. O maior conhecimento dos diversos aspectos resultou na divisão das diretrizes em duas partes, sendo nessa primeira parte abordados a definição, a classificação e o diagnóstico.
Descritores: Angioedema, angioedema hereditário, diagnóstico, classificação, diagnóstico diferencial.
ANGIOEDEMA HEREDITÁRIO: UM OLHAR PARA O MELHOR CUIDADO
Nas últimas décadas houve avanço importante no conhecimento sobre a fisiopatologia e acesso ao diagnóstico molecular do angioedema, o que permitiu a identificação de novas formas associadas ao angioedema hereditário (AEH)1. Além disso, estes avanços possibilitaram o desenvolvimento de novas drogas, eficazes e seguras, para o tratamento do AEH. Como consequência, houve maior divulgação da doença, o que resultou em maior número de pacientes identificados, embora as pesquisas nacionais ainda mostrem um atraso importante no diagnóstico do AEH, resultando em maiores morbidade e mortalidade2.
O caráter imprevisível e potencialmente fatal do AEH impacta negativamente a qualidade de vida dos indivíduos afetados e de seus familiares2-4. Embora essa condição se caracterize pela presença de sintomas apenas nos períodos de crise de angioedema, outros aspectos influenciam a qualidade de vida que estão presentes nos períodos assintomáticos, enfatizando a necessidade de suporte contínuo aos indivíduos afetados3. Portanto, o apoio e orientações fornecidos aos pacientes e familiares pela Associação Brasileira de Portadores de AEH (ABRANGHE), contribuem para minimizar o ônus e difundir conhecimento sobre a doença.
Nesse contexto, as primeiras diretrizes brasileiras para o diagnóstico e tratamento do angioedema hereditário foram elaboradas por especialistas da Associação Brasileira de Alergia e Imunologia (ASBAI) em 2010 e atualizadas em 2017, com a ativa presença do Grupo de Estudos Brasileiro em Angioedema Hereditário (GEBRAEH).
A atualização das diretrizes brasileiras visa difundir o conhecimento sobre o AEH, estabelecer normas referentes ao seu diagnóstico e tratamento no Brasil, seguindo as melhores evidências e recomendações das diretrizes internacionais, com um olhar para o melhor cuidado dos pacientes. As diretrizes internacionais mais recentes indicam que os objetivos principais do tratamento do AEH devem ser alcançar o controle total da doença e proporcionar uma vida normal ao paciente5. Além disso, desde a última atualização das diretrizes, em 2017, novos tratamentos foram desenvolvidos, assim como novas formas de administração dos medicamentos já existentes foram aprovadas pela ANVISA, justificando a necessidade da presente atualização.
As diretrizes brasileiras para o diagnóstico e tratamento do angioedema hereditário de 2022 serão apresentadas em duas partes. Na primeira, serão abordados a definição, a classificação e o diagnóstico do AEH e, na segunda parte, a abordagem terapêutica. Foi realizada revisão da literatura não sistemática com a seleção de consensos/diretrizes e artigos relevantes na base de dados MEDLINE empregando-se o PubMed. Além disso, pontos controversos foram debatidos entre os autores participantes.
O QUE É O ANGIOEDEMA HEREDITÁRIO?
O angioedema é um edema transitório, circunscrito, assimétrico, deformante, não inflamatório, não pruriginoso, por vezes, doloroso, localizado na camada subcutânea da pele e/ou na submucosa de alguns órgãos6,7.
O AEH foi descrito pela primeira vez em 1882, por Quincke, originalmente como edema "angioneurótico", por sua associação com transtornos psicológicos ou psiquiátricos8-10. Em 1888, Osler estabeleceu a sua natureza hereditária, porém, a primeira alteração bioquímica associada à doença, a deficiência do inibidor de C1 esterase (C1-INH), só foi identificada 75 anos após, quando se definiu o AEH como uma deficiência quantitativa ou qualitativa do C1 inibidor (AEH-C1-INH)11-13.
O AEH é doença genética rara, potencialmente fatal e subdiagnosticada, caracterizada por crises recorrentes de edema que podem acometer tanto a derme e o tecido subcutâneo quanto órgãos internos, predominantemente o sistema digestório e as vias respiratórias superiores14-15. Caracteriza-se por angioedema sem a presença de urticas, diferentemente do angioedema histaminérgico, onde aproximadamente 90% dos pacientes apresentam essas lesões cutâneas16. Aproximadamente um terço dos pacientes com angioedema recorrente sem urticas podem ter diagnóstico de AEH17-18. Atualmente dois grupos principais de AEH são reconhecidos: o angioedema com deficiência de C1-INH (AEH-C1-INH), e o AEH com C1-INH normal (AEH-nC1-INH). A prevalência média mundial do AEH-C1-INH foi estimada em aproximadamente 1:67.000 (1,5 por 100.000 habitantes), enquanto o AEH-nC1-INH é mais raro, estimando-se que ocorra em 1:400.000 indivíduos19-20.
QUAIS AS CAUSAS DO ANGIOEDEMA HEREDITÁRIO?
O C1-INH é uma glicoproteína codificada pelo gene SERPING1, localizado no cromossomo 11, e que tem mais de 700 mutações já descritas no AEH-C1-INH1. O C1-INH é membro da superfamília das serpinas ou inibidores de serinoproteases; atua como inibidor suicida que aprisiona, de modo irreversível, a proteína-alvo em um complexo inativo, altamente eficiente21,22. Variantes patogênicas no gene SERPING1 resultam na redução quantitativa da produção do C1-INH principalmente pelo hepatócito, e na diminuição de sua atividade funcional ocasionando o AEH-C1-INH tipo I, responsável por 85% dos casos. No AEH-C1-INH tipo II há a produção de uma proteína disfuncional sem alteração nos níveis quantitativos do C1-INH, identificada nos 15% dos casos restantes23-25.
O padrão de herança no AEH-C1-INH é autossômico dominante. Em 25% dos pacientes ocorre uma mutação de novo, sem história familiar evidente da doença26-28. No AEH-C1-INH, a mutação ocorre em uma das duas cópias do gene SERPING1, com raros casos de homozigose publicados. As mutações que resultam em AEH-C1-INH tipo I podem ocorrer em qualquer parte do gene SERPING1, enquanto as mutações responsáveis pelo AEH-C1-INH tipo II ocorrem no exon 8, onde se localiza a alça do centro reativo do C1-INH, originando uma proteína disfuncional28. No AEH-C1-INH tipo I, os níveis plasmáticos do C1-INH deveriam ser próximos aos 50%, porém, os pacientes com este tipo de AEH apresentam níveis que variam entre 5% e 30% dos níveis plasmáticos normais. Essa discrepância pode ser explicada pelo achado de que o C1-INH produto do gene SERPING1 mutado forma agregado com o C1-INH normal, e este agregado é retido no retículo endoplasmático, configurando um mecanismo dominante negativo24,28. A penetrância bioquímica, com alterações laboratoriais, se aproxima dos 100%, mas a expressão clínica e a gravidade da doença são altamente variáveis28.
Em 2000, foram descritos pela primeira vez pacientes e famílias com angioedema que manifestavam sintomas semelhantes aos de pacientes com AEH-C1-INH, entretanto, com níveis quantitativos e funcionais de C1-INH normais. Este tipo de AEH foi incialmente descrito como AEH tipo III, entretanto esta nomenclatura não é mais usada, e este tipo de AEH é atualmente designado como AEH com C1-INH normal (AEH-nC1-INH)29. O padrão de herança no AEH-nC1-INH também é autossômico dominante28.
QUAL É O MECANISMO ENVOLVIDO NO ANGIOEDEMA HEREDITÁRIO?
O C1 é o primeiro componente da via clássica do complemento e forma um complexo ao unir-se a uma molécula de C1q, duas de C1r e duas de C1s. Este complexo tem autoativação lenta, porém, ao se ligar a um imunocomplexo, o C1 adquire sua atividade completa. O C1-INH inibe a forma ativada do C1, estabilizando-o e diminuindo a sua ativação. Cada molécula ativada de C1r e C1s se liga irreversivelmente a uma molécula do C1-INH30,31.
Inicialmente, o C1-INH foi reconhecido apenas por sua atividade na inibição do sistema complemento, tanto nas vias clássicas como na das lectinas, sem a qual resultaria em um sistema excessivamente ativado. Posteriormente, o C1-INH também foi associado à inibição de várias proteases, incluindo a calicreína do plasma, os fatores de coagulação XII (FXII) e XI, e a plasmina. Portanto, além de inibir o sistema complemento, o C1-INH participa na regulação dos sistemas de contato e calicreína/cinina, da coagulação e de fibrinólise25,31-33. Estudos adicionais revelaram que a deficiência de C1-INH no AEH-C1-INH resulta em excesso de produção de bradicinina (BK) que se liga ao receptor B2 (BDKRB2), desempenhando papel importante no angioedema34-36. O desenvolvimento de novos tratamentos, como o antagonista do receptor B2 de BK e inibidores de calicreína, reforçou o papel da BK como principal mediador no AEH-C1-INH37,38.
Comumente usados como sinônimos, e apesar de exibirem sobreposição e interações, os termos sistemas plasmáticos de contato (SC) e calicreína-cinina (SCC) são diferentes. O SC refere-se ao sistema proteolítico iniciado pela autoativação do fator XII (FXII), enquanto o SCC é constituído pela calicreína que cliva o cininogênio de alto peso molecular (HMWK) e, com isso, libera um nonapeptídeo vasoativo, a BK39. A ativação do FXII, com geração de FXIIa, é iniciada por superfícies negativamente carregadas, ou macromoléculas. Subsequentemente, o FXIIa ativa mais FXII, em um processo de autoativação. O próximo substrato da cascata é a pré-calicreína, que será convertida em sua forma ativa, a calicreína que por sua vez degrada o HMWK, liberando a BK. Ao se ligar ao seu receptor B2, que é constitutivamente expresso em células endoteliais, a BK interfere nas junções endoteliais, aumentando a permeabilidade vascular e induzindo ao angioedema. A BK também estimula a produção de óxido nítrico pelas células endoteliais, que, consequentemente, desencadeia vasodilatação por contração do citoesqueleto. Vale salientar que a ativação do FXII ocorre próximo da parede endotelial, determinando a ativação da cascata que resulta na produção de BK que se liga, de forma parácrina, ao receptor B2 no endotélio. Adicionalmente, a calicreína ativa diretamente o FXII e também atua no sistema fibrinolítico, convertendo o plasminogênio em plasmina que, por sua vez, ativa mais FXII, formando um ciclo de retro ativação (Figura 1)22,25,31,40-43.
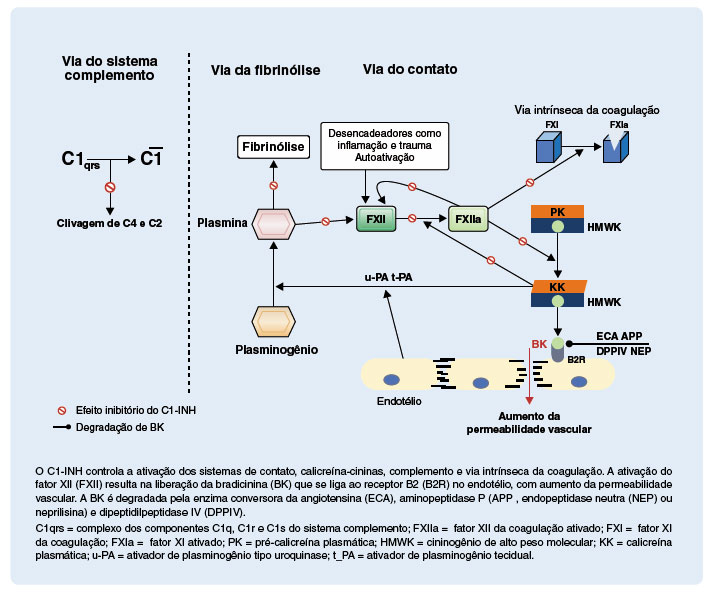
Figura 1
Locais de ação do inibidor de C1 (C1-INH) nos sistemas de contato, calicreína-cininas, complemento e via intrínseca da coagulação
A BK no plasma tem meia-vida extremamente curta, pois é prontamente degradada por várias peptidases (Figura 1). A enzima conversora da angiotensina (ECA) é a peptidase mais importante para a degradação da BK. A dipeptidil peptidase IV (DPPIV , endopeptidase neutra (NEP) ou neprilisina) e a aminopeptidase P (APP) são outras peptidases que atuam no mesmo processo. A BK é transformada por estas enzimas em des-Arg-BK, que é a sua forma inativa40,44.
Dentre os sistemas inibidos pelo C1-INH, os SC e SCC têm maior relevância para a gênese do AEH-C1-IN-H. O C1-INH inibe a autoativação do FXII em FXIIa, a conversão de pré-calicreína em calicreína, a ativação do FXII pela calicreína e a clivagem proteolítica do HMWK com a liberação da BK. Com todas estas etapas ineficientemente inibidas no AEH-C1-INH, ocorrerá liberação exagerada de BK25,30.
Nos últimos anos, o papel do endotélio local vem ganhando importância na tentativa de explicar a natureza local do angioedema. Em resposta a vários estímulos, como infecção, trauma e estresse, o endotélio libera substâncias vasoativas que modulam tanto a vasodilatação quanto a vasoconstricção, assim como a permeabilidade vascular. A pré-calicreína plasmática circula em complexo com o HMWK e este complexo pode ser recrutado para a superfície do endotélio. Assim, o endotélio desempenha papel fundamental na indução da crise de angioedema. O mecanismo responsável pela indução deste processo numa determinada parte do organismo, e não em outra, e o fato da crise ser localizada e não se tornar sistêmica ainda é desconhecido. Além disso, já foi demonstrado que o C1-INH se liga a moléculas de adesão na parede do endotélio, aumentando assim o seu contato com o SC e SCC, além do sistema complemento, tornando a sua ação inibitória mais eficiente. Em resumo, o endotélio, pela ação de alguns estímulos, pode se tornar localmente ativado iniciando o processo que culmina na liberação e ação compartimentalizada da BK25.
No AEH-nC1-INH, mutações no gene F12 codificador do FXII foram descritas em uma parcela das famílias de pacientes, e este tipo de AEH foi designado como AEH-FXII45. Dentre as quatro mutações no gene F12 causadoras de AEH-FXII, todas localizadas no exon 9, a mutação missense c.983C>A, que leva à substituição do amino ácido treonina pela lisina na posição 328 da proteína FXII (p.Thr328Lys), tem sido a mais frequentemente encontrada20. As variantes patogênicas no gene F12 tornam o FXII mais susceptível à ativação pela plasmina e outras proteases20,28,46. A trombina que é gerada após o trauma pode ativar o FXII e explicar o angioedema após esse estímulo47. Como já foi descrito, o FXII tem papel central nas fases iniciais da ativação dos SC e SCC, e no aumento da liberação de BK25.
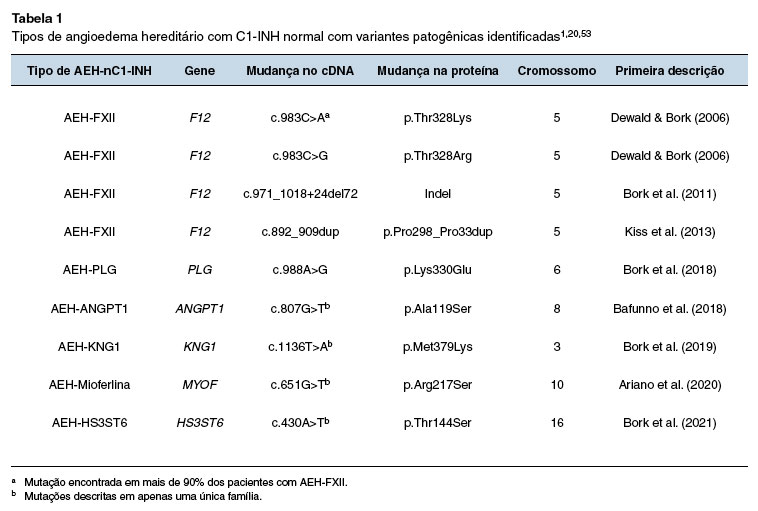
No entanto, nem todos os pacientes com AEH-nC1-INH apresentavam mutação do F12, ou seja, a maioria permanecia com o AEH de causa desconhecida - unknown (AEH-U)48. Com o advento de novas tecnologias de sequenciamento completo do exoma, mutações em outros cinco genes além do F12 foram descritas em famílias de pacientes com AEH-nC1-INH1 (Tabela 1). Uma mutação em heterozigose e transmissão autossômica dominante no gene do plasminogênio (PLG) foi descrita e, até o momento, o mecanismo de angioedema ainda não está esclarecido49. No mesmo ano, uma mutação no gene da angiopoietina 1 (ANGPT1) foi identificada. Esta mutação amplia o espectro fisiopatológico do angioedema, visto que envolve um gene não relacionado aos SC e SCC. A mutação do ANGPT1 resulta na síntese de quantidades diminuídas da ANGPT1 no plasma e numa menor ligação ao seu receptor Tie2 (tunica interna endothelial cell kinase 2). A ligação da ANGPT1 ao Tie2 é importante para a estabilização e a diminuição da permeabilidade vascular50. Uma outra mutação no gene do cininogênio 1 (KNG1) foi descrita em 2019 e o mecanismo do angioedema ainda é desconhecido, mas pode estar relacionado ao processo da formação da BK51. Posteriormente, uma mutação com possível ganho de função, no gene da mioferlina (MYOF) foi associado a um novo subtipo de AEH-nC1-INH. A mioferlina é uma proteína de membrana das células endoteliais e modula a transdução de sinal via fator de crescimento endotelial vascular (VEGF). A interação da mioferlina com as vias de sinalização do VEGF pode estar relacionada à liberação do óxido nítrico que, por sua vez, é um importante mediador da permeabilidade vascular52. A mutação mais recentemente descrita, foi a do gene do heparan sulfato-glucosamina 3-O-sulfotransferase 6 (HS3ST6). Suspeita-se que esta mutação leve à síntese incompleta do heparan sulfato, afetando a estrutura dos proteoglicanos, e a consequente alteração na interação do HMWK com as células endoteliais53.
O entendimento da influência do estrogênio, endógeno e/ou exógeno sobre o AEH-C1-INH, tanto nos tipos I e II como no AEH-FXII, ainda é incompleto. O AEH-C1-INH é negativamente impactado pelo estrogênio, e o AEH-FXII já foi considerado estrogênio-dependente, visto que alguns pacientes com mutação em F12 têm manifestações clínicas apenas após a gestação ou com o uso de anticoncepcionais contendo este hormônio54-56. O estrogênio estimula a liberação de algumas citocinas e da proteína do choque térmico, denominada Hsp90, que nas células endoteliais pode converter a pré-calicreína em calicreína, e esta, clivar o HMWK, liberando BK. Adicionalmente, a calicreína ativa o FXII diretamente ou induzindo a degradação do plasminogênio em plasmina, que por sua vez ativa o FXII. Portanto, o estrogênio, ao estimular a liberação da Hsp90, induz a ativação do FXII, principalmente no AEH-FXII57. A região promotora do F12 contém um elemento de resposta ao estrogênio e foi demonstrado o aumento da transcrição de mRNA de FXII em resposta ao hormônio. É provável que o estrogênio também contribua para o aumento da expressão do receptor B2 da BK. A ação da BK é mediada pelo óxido nítrico, e o estrogênio é um regulador da liberação dessa substância, contribuindo para o angioedema mediado pela BK. Por fim, o estrogênio pode diminuir a degradação da BK ao interferir na atividade da enzima conversora da angiotensina33,58,59.
Portanto, até o momento foram descritas mutações em sete genes diferentes nos pacientes com AEH, sendo as mais frequentes no gene SERPING1 seguidas por mutações no gene F12 relacionadas ao AEH-C1-INH e AEH-FXII, respectivamente20. Outras mutações identificadas em pacientes e famílias com AEH envolvem genes que codificam proteínas que participam das vias de produção de BK, como SERPING1, F12, KNG1 e possivelmente PLG (sistema fibrinolítico)20. Mutações em genes envolvidos na regulação da permeabilidade vascular no nível do endotélio, como ANGPT1 e MYOF, ou na regulação endotelial do sistema de cininas, como o HS3ST6, têm também sido descritas em pacientes com AEH-nC1-INH, revelando novas vias patogênicas que podem vir a serem alvos terapêuticos1,20,53. Deste modo, o angioedema hereditário também pode ser classificado baseado nos novos mecanismos descritos:
- Angioedema por bradicinina: AEH-C1-INH (tipos I e II), AEH-FXII, AEH-PLG, AEH-KNG;
- Angioedema por disfunção do endotélio vascular: AEH-ANGPT1, AEH-MYOF e AEH-HS3ST6.
QUAIS AS MANIFESTAÇÕES CLÍNICAS TÍPICAS DE ANGIOEDEMA HEREDITÁRIO?
Características gerais
Os sintomas do AEH podem ter início em qualquer idade, entretanto, na maioria dos pacientes iniciam-se na primeira ou segunda década de vida. Os estudos demonstram início dos sintomas em 75% dos pacientes com AEH-C1-INH até os 15 anos de idade2,26,60-64. De modo geral, em 50% dos casos de AEH-C1-INH, o início dos sintomas ocorre em torno dos 10 anos de idade, com aumento da frequência e gravidade das crises na puberdade65. No AEH-nC1-INH, a maioria dos casos é desencadeada na adolescência, como relatado em coorte brasileira, na qual 72% dos pacientes (n = 197) apresentaram a primeira crise entre a segunda e a terceira décadas de vida66,67.
O AEH manifesta-se por episódios recorrentes e imprevisíveis de angioedema, em qualquer parte do corpo68-70. A frequência e a gravidade das crises de AEH é variável entre os pacientes e ao longo da vida em um mesmo paciente5,68,69,71,72. É descrito que 5% dos indivíduos com AEH são assintomáticos, e 25% desenvolvem sintomas esporádicos69,73-75. A frequência das crises é individual e varia de episódios esporádicos a mais de uma crise por semana. Esta ampla variação na expressão fenotípica não tem correlação com as concentrações plasmáticas de C1-INH, e provavelmente outros fatores genéticos e/ou fatores ambientais podem influir na frequência das crises76.
Observa-se um pico de sintomas entre 12 e 24 horas, regredindo espontaneamente em dois a cinco dias. A instalação do edema costuma ser lenta e gradual, e ocorre geralmente em torno de oito horas. Entretanto, em locais como abdome e laringe, o angioedema pode instalar-se mais rapidamente68-70. No angioedema hereditário não ocorrem urticas e não há resposta ao tratamento com anti-histamínicos, corticosteroides e adrenalina6,7.
Os AEH-C1-INH dos tipos I e II não diferem quanto aos sintomas clínicos. Embora o AEH acometa ambos os sexos, tende a ser mais grave e frequente na mulher, pelo papel do estrogênio na patogênese da doença. A apresentação clínica do AEH-nC1-INH é similar ao AEH-C1-INH, porém os sintomas são menos frequentes, e outras diferenças também são descritas66,72.
O curso da doença tende a ser mais grave quanto mais precoce é o início dos sintomas69,77. Da mesma forma, observa-se piora na frequência e gravidade das crises após a puberdade, tanto em mulheres quanto em homens65. Em alguns pacientes de idade mais avançada os sintomas tornam-se mais leves, entretanto, as crises de angioedema raramente cessam completamente65,68.
Fatores desencadeantes
Os episódios de angioedema podem ocorrer espontaneamente, mas em até 91% dos casos é induzido por fatores físicos, psicológicos, infecciosos, medicamentosos ou hormonais5,78-80. O estresse emocional é relatado pelos pacientes com AEH-C1-INH como o mais frequente fator desencadeante de crises80,81. O trauma mecânico, mesmo que leve, é o segundo desencadeante em frequência no AEH-C1-INH, e o angioedema inicia-se caracteristicamente na área traumatizada82. O angioedema pode ser desencadeado por procedimentos odontológicos, cirúrgicos ou diagnósticos, ocorrendo geralmente em torno de 4 a 36 horas após a intervenção83,84. As infecções em geral, em especial as virais, são consideradas como um gatilho relevante de crises de AEH, principalmente em crianças82,85. Situações em que há elevação dos níveis de estrogênio, como o uso de contraceptivos orais, terapia de reposição hormonal durante a menopausa, gravidez e menstruação são de risco potencial para desencadeamento de crises no AEH-C1-INH86.
Diversas drogas que interferem no metabolismo da BK têm sido descritas como associadas ao maior risco de crises de angioedema. Na grande maioria dos casos, o mecanismo envolve inibição da degradação da BK, resultando em elevação de seu nível sérico e, consequentemente, angioedema40. Os medicamentos utilizados no tratamento da hipertensão arterial com ação no sistema renina-angiotensina-aldosterona (SRAA), como os inibidores da enzima conversora da angiotensina (iECA) e, com menor risco, os bloqueadores do receptor da angiotensina II (BRAs), foram associados ao angioedema adquirido, entretanto, podem também desencadear AEH40,87.
Do mesmo modo, as gliptinas que inibem a enzima DPPIV usados como hipoglicemiantes orais, diminuem o catabolismo da BK e consistem em potenciais gatilhos para o desenvolvimento de crises40,87. Os inibidores da neprilisina, uma outra classe de medicamentos utilizada no tratamento da hipertensão arterial e insuficiência cardíaca, tal como o sacubitril, podem causar angioedema, especialmente quando usados em combinação com inibidores do SRAA40,87,88. Os inibidores da proteína intracelular mTOR, imunossupressores utilizados no tratamento do câncer, representam risco adicional para pacientes com AEH-C1-INH89.
Outros desencadeantes menos frequentes de crises de angioedema são descritos pelos pacientes, tais como exposição a temperaturas extremas, consumo de álcool, ingesta de alguns alimentos, e fadiga80,90,91.
Pródromos
Os pródromos são relatados em vários pacientes com AEH-C1-INH, precedendo a crise em uma a 24 horas81,92,93. Um terço dos pacientes apresenta lesões cutâneas maculares, eritemato-serpiginosas, fugazes e não pruriginosas, geralmente no tronco e membros, conhecidas como eritema marginatum, ou também, eritema serpiginoso94. Em crianças, o eritema marginatum é descrito como um fenômeno independente, sem angioedema subsequente e, frequentemente, como manifestação inicial do AEH-C1-INH95. Sintomas inespecíficos, como astenia, sede, fome, náusea, fadiga mental, mudanças de humor, depressão, ansiedade, irritabilidade, agressividade, dores musculares, formigamento ou sensação de aperto na área que será afetada, além de sintomas semelhantes aos da gripe, também foram relatados como sintomas prodrômicos81,93,94.
O eritema marginatum não foi relatado como pródromo em pacientes com AEH-nC1-INH, entretanto outros sintomas inespecíficos como fadiga, dor torácica e palpitações foram observados em alguns casos20. Alguns pacientes com AEH-nC1-INH apresentam lesões cutâneas que lembram equimose, porém, há poucos casos descritos96-99.
As manifestações prodrômicas possibilitam o início precoce do tratamento em caso de crise e, assim, diminuir a morbimortalidade associada ao AEH-C1-INH. Entretanto, as queixas são muito variáveis e não há, até o momento, evidência científica do seu valor preditivo92,93. A presença do eritema marginatum, pródromo mais característico do AEH-C1-INH, tem sido associada a atraso no diagnóstico da doença, pois, é frequentemente confundido com urticária100.
Localização
Os três locais de acometimento mais característicos do AEH são: subcutâneo, abdome e laringe2,68-70. O acometimento subcutâneo é o mais frequente, afetando 95% dos pacientes com AEH-C1-INH, destacando-se as extremidades, genitália e face como os locais mais comumente envolvidos. O abdome é o segundo local de acometimento mais comum, ocorrendo em até 93% dos casos. Nas crises, vários locais podem ser acometidos simultaneamente, característica que distingue o AEH de outras causas de angioedema sem urticária2,6,68-70.
A dor abdominal decorrente do edema de alças intestinais pode ser intensa e espasmódica, com duração de muitas horas a vários dias. Frequentemente, estes sintomas podem ser confundidos com abdome agudo cirúrgico, resultando em apendicectomias e laparotomias exploradoras desnecessárias em até um terço dos pacientes, tanto no AEH-C1-INH quanto no AEH-nC1-INH101,102. O edema da mucosa do trato gastrointestinal pode causar compressão do lúmen e quadro clínico de íleo paralítico temporário, provocando náusea e vômito. A diarreia aquosa devido ao acúmulo de líquido na luz do intestino edematoso também é comum. O extravasamento de líquido para a cavidade peritoneal pode resultar em ascite com aumento do volume do abdome, frequentemente identificada pela ultrassonografia como líquido na cavidade abdominal103. A hemoconcentração, a hipotensão arterial e até mesmo choque hipovolêmico podem ocorrer nesses pacientes, secundariamente à perda substancial de fluidos para o interstício ou cavidade69.
O acometimento da laringe, mais precisamente na região supraglótica, ocorre em 50% dos pacientes e, pelo menos, um episódio ocorre durante a vida do paciente2,68-70. O edema de laringe é mais frequente entre os 11 e 45 anos de idade, e raro antes do terceiro ano de vida104,105. Apesar de menos frequente que os sintomas cutâneos e abdominais, o edema laríngeo é potencialmente fatal, particularmente nos pacientes não tratados. Outro aspecto importante é que pode haver edema de regiões acima da laringe, tais como a base da língua e orofaringe, que pode também impedir a passagem do ar e resultar em asfixia e, desse modo, o termo edema de vias respiratórias superiores, ao invés de edema de laringe, tem sido utilizado73,78,106. Foi descrita alta frequência de edema de língua em pacientes com mutação no gene que codifica o plasminogênio (AEH-PLG)20. O tempo de instalação do angioedema laríngeo mais frequente é de aproximadamente oito horas, no entanto, pode ter início súbito e ocasionar obstrução aguda das vias respiratórias104,105. Vale salientar que pacientes com edema facial são considerados de risco para edema de vias respiratórias superiores. Apesar de ser menos frequente que os sintomas cutâneos e abdominais, o edema laríngeo letal pode ser a primeira apresentação do AEH-C1-INH104,105.
Manifestações clínicas incomuns do AEH-C1-INH, como bolhas lineares, cefaleia intensa, disúria e pancreatite aguda também foram descritas na literatura médica107-109.
QUAIS EXAMES CONFIRMAM O DIAGNÓSTICO?
Todo indivíduo com suspeita clínica de AEH ou que tem histórico familiar de sintomas semelhantes, deve realizar os exames laboratoriais para confirmação do AEH-C1-INH. A dosagem do nível sérico de C4 pode ser utilizada como triagem de AEH (Figura 2). Na deficiência quantitativa ou disfunção do C1-INH, a estabilização do complexo C1 está baixa, se tornando parcialmente ativado e o C4, seu substrato preferencial, depletado. Na maioria dos pacientes com AEH-C1-INH tipos I e II, o C4 está continuamente diminuído no plasma, porém há a possibilidade de níveis de C4 normal79. Nessas situações, é recomendada a dosagem de C4 no período de crise, caso a história clínica seja sugestiva e a análise dos níveis de C1-INH não seja disponível5,110. A avaliação do C1-INH quantitativa (por imunodifusão radial ou turbidimetria/nefelometria) e funcional (por ensaio cromogênico) são recomendadas para o diagnóstico definitivo. A maior parte dos pacientes (85%) apresenta C1-INH quantitativo abaixo de 50% da faixa de normalidade, estabelecendo-se o diagnóstico de AEH-C1-INH tipo I79. Quando a concentração de C1-INH se encontra normal (ou mesmo alta, em alguns casos), o teste funcional de C1-INH é essencial. O diagnóstico de AEH-C1-INH tipo II é caracterizado por C1-INH quantitativo normal ou elevado e atividade funcional de C1-INH reduzida, e correspondendo a aproximadamente 15% dos pacientes com AEH-C1-INH111. No Brasil, o exame de C4 é amplamente disponível em laboratórios de análises clínicas, enquanto os outros testes são realizados apenas em laboratórios mais especializados. É importante ressaltar que a coleta e manipulação das amostras podem ser fatores limitantes para o amplo acesso à avaliação do C1-INH, pois pode ocorrer a degradação e consumo dos componentes do complemento com muita facilidade112. Desta forma, as dosagens bioquímicas necessárias ao diagnóstico do AEH-C1-INH, especialmente a avaliação funcional de C1-INH, pode gerar resultados falso-positivos, sendo aconselhada a realização de ao menos duas dosagens coletadas em dias diferentes5,111,113.
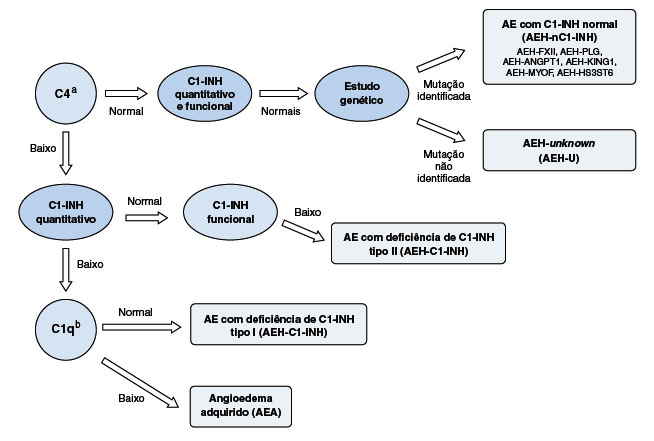
Figura 2
Algoritmo do diagnóstico do angioedema hereditário1,5,14,72
a Se C4 normal, repetir durante a crise de angioedema.
b Solicitar na dependência da história clínica.
AE = angioedema, AEH = angioedema hereditário, AEA = angiodema adquirido, AEH-U = angioedema hereditário de causa desconhecida, AEH-FXII = angioedema hereditário por mutação no gene do Fator XII, AEH-PLG = angioedema hereditário por mutação no gene do plasminogênio, AEH-ANGPT1 = angioedema hereditário por mutação no gene da angiopoietina 1, AEH-KNG1 = angioedema hereditário por mutação no gene do cininogênio 1, AEH-MYOF = angioedema hereditário por mutação no gene da mioferlina, AEHHS3ST6 = angioedema hereditário por mutação no gene do heparan sulfato 3OST6.
No angioedema adquirido por deficiência do C1-INH (AEA-C1-INH), as concentrações de C4 e de C1-INH, assim como a avaliação funcional, podem estar reduzidos. Neste caso, a dosagem de C1q deve ser realizada e encontra-se reduzida em aproximadamente 70% dos casos. Características clínicas como sintomas de início mais tardio na idade adulta e ausência de história familiar de angioedema recorrente sugerem o diagnóstico114,115. Ainda, considerando-se que um percentual dos pacientes com AEA-C1-INH pode apresentar níveis plasmáticos normais de C1q, a avaliação genética do gene SERPING1 é recomendada para o diagnóstico diferencial. Em casos com mutações de novo ou história clínica questionável, a avaliação genética também pode ser necessária116.
Em crianças menores de um ano de idade, os níveis plasmáticos do C1-INH podem estar abaixo dos valores considerados normais por imaturidade imunológica, recomendando-se a análise genética de SERPING1 para auxiliar no diagnóstico do AEH-C1-INH77,117.
A genotipagem de SERPING1 pode ser realizada por sequenciamento de Sanger ou sequenciamento de nova geração, devendo cobrir os oito exons do gene, incluindo seus sítios de splicing. Na ausência de variantes patogênicas, deve-se avaliar a presença de grandes deleções e inserções utilizando técnicas como multiplex ligation-dependent probe amplification (MLPA) ou PCR long-range, embora esses testes não sejam amplamente disponíveis28. A avaliação de patogenicidade de novas variantes identificadas no SERPING1 deve seguir as diretrizes internacionais estabelecidas pelo American College of Medical Genetics and Genomics (ACMG) para o AEH20,118. Embora não seja fundamental para o diagnóstico do paciente sintomático, a determinação das mutações causadoras do AEH-C1-INH auxilia na triagem familiar e prevenção precoce, inclusive em portadores assintomáticos.
Para os casos de suspeita de AEH e resultados consistentes e normais de C4 e C1-INH, deve-se investigar o AEH-nC1-INH, para o qual não há marcadores bioquímicos disponíveis, e a única alternativa é o diagnóstico genético1,20.
Em pacientes com AEH, estima-se que o atraso para o diagnóstico ainda é elevado, e os estudos nacionais documentam que esta demora no diagnóstico é variável entre 14 e 18 anos2,26,60-64.
QUAIS SÃO OS CRITÉRIOS DIAGNÓSTICOS DO AEH?
Alguns critérios para padronizar o diagnóstico de AEH têm sido propostos (Tabela 2)72. Dentre eles, alguns são requeridos para o diagnóstico, ao passo que outros se constituem como forte indício, mas não são necessários sendo, portanto, critérios de suporte. Por exemplo, a detecção de mutação no gene SERPING1 no AEH-C1-INH não é necessária para o diagnóstico, sendo, dessa forma, um critério de suporte. A característica do angioedema subcutâneo não inflamatório com duração superior a 12 horas e a presença de dor abdominal de etiologia orgânica indefinida, com duração superior a seis horas, além do edema de laringe, são características importantes no AEH14.
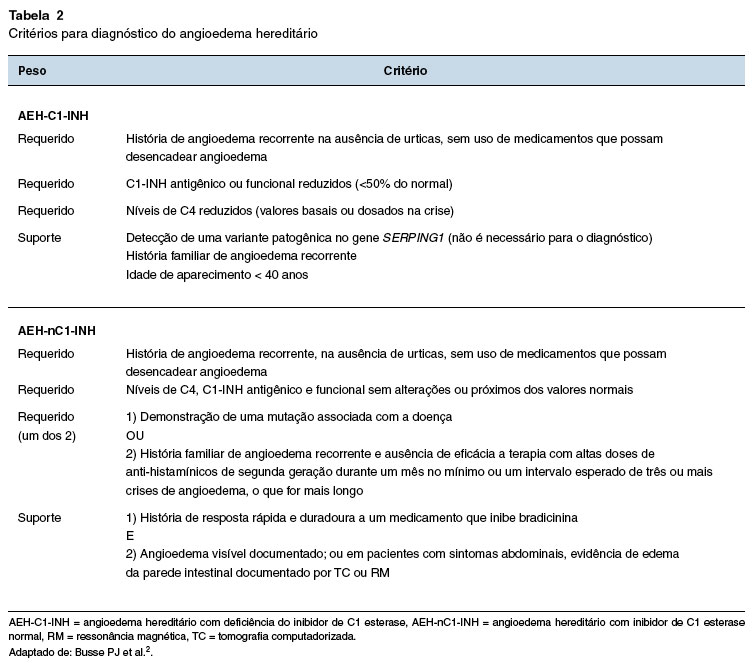
Esses critérios não são absolutos e a história clínica é preponderante, especialmente em localidades onde os exames laboratoriais não estão disponíveis. No AEH-nC1-INH, um teste terapêutico pode ajudar no estabelecimento do diagnóstico72.
Na Figura 3, sugerimos uma lista de sinais de alerta e um acrônimo para estimular a suspeita diagnóstica e promover a conscientização sobre o AEH-C1-INH.
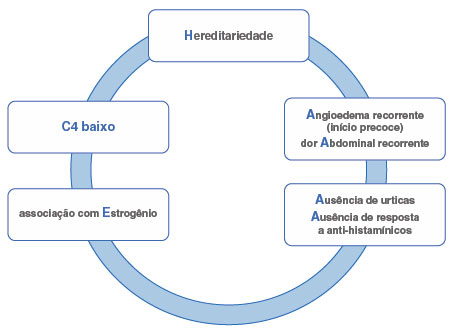
Figura 3
Sinais de alerta para o diagnóstico de AEH-C1-INH14
HAAAAE = Hereditariedade, Angioedema recorrente, dor Abdominal recorrente, Ausência de urticas, Ausência de resposta a anti-histamínicos, associação com Estrogênio. Adaptado de: Giavina-Bianchi P, et al.14.
O QUE NÃO É ANGIOEDEMA HEREDITÁRIO?
Dois mecanismos fisiopatológicos principais de angioedema são descritos: por ativação de mastócitos e/ou basófilos, resultando na liberação de histamina e outros mediadores (angioedema histaminérgico); e por excesso de BK (angioedema mediado pela bradicinina ou não-histaminérgico), como visto no AEH-C1-INH, AEA-C1-INH, e angioedema induzido por iECA ou por gliptinas, medicamentos envolvidos no metabolismo de BK (Figura 4)8,17,119. Portanto, os principais diagnósticos diferenciais do AEH são os outros tipos de angioedema, principalmente aqueles com apresentação crônica ou recorrente. O conhecimento sobre os mecanismos fisiopatológicos, características clínicas e resposta aos fármacos utilizados na vigência da crise contribuem para a suspeição de outras causas de angioedema. Além de aspectos clínicos, a avaliação laboratorial auxilia na discriminação entre o angioedema mediado por histamina e o mediado por bradicinina (Tabela 3).
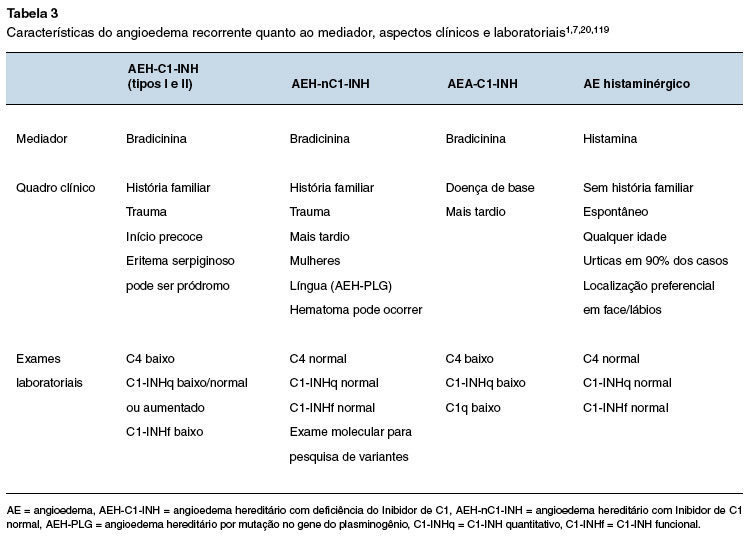

Figura 4
Classificação do angioedema1,7,8, 17,119
O tipo mais frequente de angioedema recorrente é o histaminérgico, que apresenta algumas características que o diferenciam do AEH, incluindo a presença de urticas, melhora com anti-histamínicos e desencadeamento dos sintomas pelo uso de anti-inflamatórios não hormonais (AINEs). No entanto, o angioedema histaminérgico pode se apresentar sem urticas, e os AINEs estão entre as principais causas de angioedema, mesmo naqueles pacientes que não apresentam urticária9. As diretrizes atuais para o tratamento de angioedema/urticária crônica espontânea destacam o fato de que alguns pacientes não responderão a doses convencionais de anti-histamínicos e podem necessitar de aumento de dose, atingindo até quatro vezes as doses diárias habitualmente recomendadas para controlar os sintomas. Portanto, para confirmar ou descartar a natureza histaminérgica do angioedema, um teste terapêutico com anti-histamínicos, usando quatro vezes a dose recomendada, por um período de tempo de aproximadamente seis semanas, é suficiente para avaliar sua resposta ao tratamento. Foi demonstrada a segurança do aumento da dose de anti-histamínicos, incluindo bilastina, cetirizina, levocetirizina, desloratadina, ebastina, fexofenadina e rupatadina16,120. Embora o angioedema mediado por BK seja menos frequente, o risco de mortalidade nesse tipo de angioedema é 45 vezes maior do que o do angioedema histaminérgico121.
Em relação às formas adquiridas de angioedema mediado por BK, é muito importante perguntar ao paciente sobre o uso de iECA. Como a ECA é a principal enzima envolvida na degradação da BK, sua inibição leva ao aumento das concentrações séricas deste mediador, e pode causar angioedema. Até 0,7% dos indivíduos que usam iECA apresentam angioedema recorrente, com aumento do risco entre os afrodescendentes, fumantes, idosos e no sexo feminino17,44. O angioedema induzido por iECA envolve mais frequentemente a face, língua, orofaringe e laringe, no entanto foram relatados casos esporádicos de episódios abdominais. O tempo médio para o início dos sintomas de angioedema é de 1,8 anos, entretanto os sintomas ocorrem em 25% dos casos no primeiro mês do uso da medicação. Podem, ainda, ocorrer até 10 anos após a introdução do tratamento122. O iECA deve ser interrompido em todos os pacientes com angioedema recorrente, mesmo se o angioedema foi desencadeado após vários anos de uso do medicamento. Embora as crises de angioedema induzidos por iECA possam assemelhar-se àquelas de AEH, os pacientes apresentarão níveis normais de C4 e de C1q, além de níveis quantitativos e/ou funcionais normais de C1-INH (Tabela 4). Mais raramente, os bloqueadores dos receptores da angiotensina II (BRA) e as gliptinas podem induzir angioedema123.
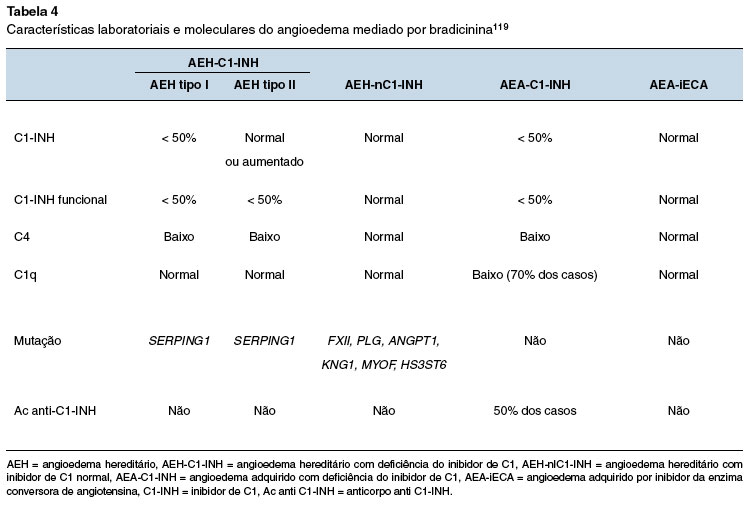
O AEA-C1-INH é um tipo de angioedema ainda mais raro que o AEH, com prevalência estimada de 1,5:1.000.000 indivíduos, sem herança genética114,115. Nesse tipo de angioedema, o aparecimento dos sintomas ocorre mais tardiamente, não existe história familiar de angioedema e a doença deve-se ao consumo do C1-INH ou à produção de autoanticorpos neutralizantes do C1-INH, associados com doenças linfoproliferativas ou doenças autoimunes, respectivamente. Como consequência, a atividade do C1-INH é baixa, o sistema complemento está ativado e o C1q geralmente reduzido, uma característica particular que pode ajudar no diagnóstico diferencial. Além da função do C1-INH abaixo de 50% do normal, os níveis de antígenos de C1-INH são geralmente reduzidos, embora a presença do C1-INH clivado possa resultar em níveis antigênicos de C1-INH normais em cerca de 20% dos pacientes. Como há grande sobreposição dos AEA-C1-INH associados a autoanticorpos e às doenças linfoproliferativas, sugere-se sua classificação como uma mesma doença124,125.
O angioedema não histaminérgico idiopático deve ser considerado quando não há hereditariedade, todas as causas conhecidas de angioedema foram excluídas e os sintomas persistem apesar do tratamento com altas doses (até quatro vezes a dose padrão) de anti-histamínicos não sedantes de segunda geração17. Há evidência de que a BK possa ser o mediador envolvido no angioedema não histaminérgico idiopático. No entanto, a evidência não é definitiva, considerando que outros mediadores vasoativos derivados de mastócitos ou outras células, incluindo cisteinil-leucotrienos, prostaglandinas e o fator ativador de plaquetas possam desempenhar um papel119. Por outro lado, o envolvimento dos mastócitos/basófilos não exclui a participação da BK, pois há evidências de que os mastócitos podem aumentar a permeabilidade vascular pela liberação de heparina, que, por sua vez, induz a formação de BK. Também há indícios da participação da liberação da BK na urticária crônica espontânea, com ou sem angioedema126,127. Entre os pacientes considerados como tendo angioedema não histaminérgico idiopático ainda pode haver indivíduos com AEH-nC1-INH, sem histórico familiar e sem mutação conhecida, assim como alguns pacientes com angioedema histaminérgico sem urticas e resistentes a anti-histamínicos9,16,17,119. Sendo assim, a identificação das diversas formas de angioedema mediado por bradicinina pode ser mais bem definida por meio de aspectos laboratoriais e moleculares específicos (Tabela 4).
CONSIDERAÇÕES FINAIS
O AEH é uma doença genética de herança autossômica dominante associada a angioedema recorrente que acomete o tecido subcutâneo e o tecido submucoso principalmente do trato digestivo e das vias respiratórias superiores5,71,72.
Existem sete tipos de AEH definidos por variantes genéticas patogênicas distintas: AEH-C1-INH, AEH-FXII, AEH-PLG, AEH-ANGPT1, AEH-KNG1, AEH-MYOF e AEH-HS3ST6. As mutações mais frequentes ocorrem no gene SERPING1, seguidas por mutações no geneF12 relacionadas ao AEH-C1-INH e AEH-FXII, respectivamente20,53.
Em muitos indivíduos com AEH, variantes genéticas causadoras da doença ainda não são conhecidas, e esses pacientes recebem o diagnóstico de AEH-U (AEH-unknown)1.
A bradicinina é o principal mediador associado às manifestações clínicas do AEH. A ação desse mediador ocorre devido à maior atividade do sistema de contato e sistema calicreína-cininas na maioria dos pacientes, ao passo que, em outros, foram descritas alterações no endotélio1,25,40,79.
Os sintomas são desencadeados na maioria das vezes por situações de estresse, trauma mecânico, infecções e medicamentos, particularmente os estrógenos, por suas ações de estímulo ao sistema de contato. Alguns pacientes apresentam sintomas prodrômicos5,7.
As manifestações clínicas são similares nos diversos tipos de AEH, sendo geralmente mais frequentes no AEH-C1-INH. O edema de laringe é o sintoma mais grave que, embora menos frequente, pode ser causa de óbito por asfixia. O AEH-C1-INH costuma surgir na infância, e as formas de AEH-nC1-INH em adultos20,105.
O exame de triagem inicial para o diagnóstico do AEH é a dosagem sérica de C4. A seguir a dosagem quantitativa e funcional do C1-INH deve ser realizada. Em alguns casos com suspeita do AEA-C1-INH, é necessário dosar o C1q. Na ausência de alterações do C1-INH, o estudo genético deve ser realizado principalmente na ausência de história familiar, ou para caracterizar um tipo específico de AEH-nC1-INH5,71,72.
O AEH pode ser confundido com o angioedema histaminérgico idiopático e também com o angioedema recorrente com uso de medicamentos, principalmente os iECA, ou com o AEA-C1-INH7,17.
REFERÊNCIAS
1. Veronez CL, Csuka D, Sheikh FR, Zuraw BL, Farkas H, Bork K. The expanding spectrum of mutations in hereditary angioedema. J Allergy Clin Immunol Pract. 2021;9(6):2229-34.
2. Alonso MLO, Valle SOR, Tórtora RP, Grumach AS, França AT, Ribeiro MG. Hereditary angioedema: a prospective study of a Brazilian single-center cohort. Int J Dermatol. 2020;59(3):341-4.
3. Bork K, Anderson JT, Caballero T, Craig T, Johnston DT, Li HH, et al. Assessment and management of disease burden and quality of life in patients with hereditary angioedema: a consensus report. Allergy Asthma Clin Immunol. 2021;17(1):40.
4. Caballero T, Prior N. Burden of Illness and Quality-of-Life Measures in Angioedema Conditions. Immunol Allergy Clin North Am. 2017;37(3):597-616.
5. Maurer M, Magerl M, Betschel S , Aberer W, Ansotegui IJ, Aygören-Pürsün E, et al. The international WAO/EAACI guideline for the management of hereditary angioedema - the 2021 revision and update. Allergy. 2022 jan 10; doi: 10.1111/all.15214. Online ahead of print.
6. Azmy V, Brooks JP, Hsu FI. Clinical presentation of hereditary angioedema. Allergy Asthma Proc. 2020; 41(Suppl 1):S18-S2.
7. Maurer M, Magerl M. Differences and Similarities in the mechanisms and clinical expression of bradykinin-mediated vs. mast cell-mediated angioedema. Clinic Rev Allerg Immunol. 2021;61(1):40-9.
8. Giavina-Bianchi P,Aun MV, Motta AA, Kalil J, Castells M. Classification of angioedema by endotypes. Clin Exp Allergy. 2015;45:1142-3.
9. Giavina-Bianchi P, Aun MV, Jares EJ, Kalil J. Angioedema associated with nonsteroidal anti-inflammatory drugs. Curr Opin Allergy Clin Immunol. 2016;16:323-32.
10. Quincke H. Über akutes umschriebenes Hautödem. Monatshefte Prakt Dermtol. 1882;1:129-31.
11. Donaldson VH, Evans RR. A biochemical abnormality in hereditary angioneurotic edema: absence of serum inhibitor of C'1-esterase. Am J Med. 1963;35:37-44.
12. Landerman NS, Webster ME, Becker EL, Ratcliffe HE. Hereditary angioneurotic edema. II. Deficiency of inhibitor for serum globulin permeability factor and/or plasma kallikrein. J Allergy. 1962;33:330-41.
13. Rosen FS, Pensky J, Donaldson V, Charache P. Hereditary angioneurotic edema: two genetic variants. Science. 1965;148(3672):957-8.
14. Giavina-Bianchi P, Arruda LK, Aun MV, Campos RA, Chong-Neto HJ, Constantino-Silva RN, et al. Diretrizes brasileiras para o diagnóstico e tratamento do angioedema hereditário - 2017. Arq Asma Alerg Imunol. 2017;1(1):23-48.
15. Minafra FG, Gonçalves TR, Alves TM, Pinto JA. The Mortality from Hereditary Angioedema Worldwide: a Review of the Real-World Data Literature. Clin Rev Allergy Immunol. 2021 Oct 23; doi: 10.1007/s12016-021-08897-8. Online ahead of print.
16. Zuberbier T, Abdul Latiff AH, Abuzakouk M, Aquilina S, Asero R, Baker D, et al. The International EAACI/GA²LEN/EuroGuiDerm/APAAACI Guideline for the definition, classification, diagnosis and management of urticaria. Allergy. 2021 Sep 18; doi: 10.1111/all.15090. Online ahead of print.
17. Cicardi M, Aberer W, Banerji A, Bas M, Bernstein JA, Bork K, et al. Classification, diagnosis, and approach to treatment for angioedema: consensus report from the Hereditary Angioedema International Working Group. Allergy. 2014;69(5):602-16.
18. Lumry WR, Settipane RA. Hereditary angioedema: Epidemiology and burden of disease. Allergy Asthma Proc. 2020;41(Suppl 1):S08-S13.
19. of Bradykinin-mediated angioedema: a sytematic investigation of epidemiological studies. Orphanet J Rare Dis. 2018;13(1):73.
20. Bork K, Machnig T, Wulff K, Witzke G, Prusty S, Hardt J. Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: a systematic review of qualitative evidence. Orphanet J Rare Dis. 2020;15(1):289.
21. Lucas A, Yaron JR, Zhang L, Ambadapadi S. Overview of Serpins and Their Roles in Biological Systems. Methods Mol Biol. 2018;1826:1-7.
22. Christiansen SC, Busse PJ. Hereditary Angioedema. Reply. N Engl J Med. 2020;383(4):e20.
23. Longhurst HJ, Bork K. Hereditary angioedema: an update on causes, manifestations and treatment. Br J Hosp Med. 2019;80(7):391-8.
24. Haslund D, Ryø LB, Seidelin Majidi S, Rose I, Skipper KA, Fryland T, et al. Dominant-negative SERPING1 variants cause intracellular retention of C1 inhibitor in hereditary angioedema. J Clin Invest. 2019;129(1):388-405.
25. Wu MA, Bova M, Berra S, Senter R, Parolin D, Caccia S, et al. The central role of endothelium in hereditary angioedema due to C1 inhibitor deficiency. Int Immunopharmacol. 2020;82:106304.
26. Bernstein JA. Update on angioedema: evaluation, diagnosis, and treatment. Allergy Asthma Proc. 2011;32(6):408-12.
27. Valle SOR, Flores PVG, França AT. Angioedema - conceito e classificação. In: França AT, Valle SO, eds. Urticária e Angioedema: diagnóstico e tratamento. 3ª ed. Rio de Janeiro: Revinter; 2014. p. 247-56.
28. Germenis AE, Margaglione M, Pesquero JB, Farkas H, Cichon S, Csuka D, et al. International consensus on the use of genetics in the management of hereditary angioedema. J Allergy Clin Immunol Pract. 2020;8(3):901-11.
29. Bork K, Barnstedt SE, Koch P, Traupe H. Hereditary angioedema with normal C1-inhibitor activity in women. Lancet. 2000;356(9225):213-7.
30. Kaplan AP, Joseph K. Complement, Kinins, and Hereditary Angioedema: Mechanisms of Plasma Instability when C1 Inhibitor is Absent. Clin Rev Allergy Immunol. 2016;51(2):207-15.
31. Kaplan AP, Joseph K. Pathogenesis of Hereditary Angioedema: The Role of the Bradykinin-Forming Cascade. Immunol Allergy Clin North Am. 2017;37(3):513-25.
32. Bork K, Witzke G, Artmann K, Benes P, Bockers M, Kreuz W. Interaction between C1-INA, coagulation, fibrinolysis and kinin system in hereditary angioneurotic edema (HANE) and urticaria. Arch Dermatol Res. 1984;276(6):375-80.
33. Walford HH, Zuraw BL. Current update on cellular and molecular mechanisms of hereditary angioedema. Ann Allergy Asthma Immunol. 2014;112(5):413-8.
34. Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE III. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. 2002;109(8):1057-63.
35. Nussberger J, Cugno M, Amstutz C, Cicardi M, Pellacani A, Agostoni A. Plasma bradykinin in angio-oedema. Lancet. 1998;351(9117):1693-7.
36. Nussberger J, Cugno M, Cicardi M. Bradykinin-mediated angioedema. N Engl J Med. 2002;347(8):621-2.
37. Cicardi M, Levy RJ, McNeil DL, Li HH, Sheffer AL, Campion M, et al. Ecallantide for the treatment of acute attacks in hereditary angioedema. N Engl J Med. 2010;363(6):523-31.
38. Cicardi M, Banerji A, Bracho F, Malbran A, Rosenkranz B, Riedl M, et al. Icatibant, a new bradykinin-receptor antagonist, in hereditary angioedema. N Engl J Med. 2010;363(6):532-41.
39. Schmaier AH. The contact activation and kallikrein/kinin systems: pathophysiologic and physiologic activities. J Thromb Haemost. 2016;14(1):28-39.
40. Cicardi M, Zuraw BL. Angioedema Due to Bradykinin Dysregulation. J Allergy Clin Immunol Pract. 2018;6(4):1132-41.
41. Elliott DF, Horton EW, Lewis GP. Actions of pure bradykinin. J Physiol. 1960;153(3):473-80.
42. Rocha e Silva M, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am J Physiol. 1949;156(2):261-73.
43. Venema VJ, Marrero MB, Venema RC. Bradykinin-stimulated protein tyrosine phosphorylation promotes endothelial nitric oxide synthase translocation to the cytoskeleton. Biochem Biophys Res Commun. 1996;226(3):703-10.
44. Montinaro V, Cicardi M. ACE inhibitor-mediated angioedema. Int Immunopharmacol. 2020;78:106081.
45. Dewald G, Bork K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem Biophys Res Commun. 2006;343(4):1286-9.
46. de Maat S, Bjorkqvist J, Suffritti C, Wiesenekker CP, Nagtegaal W, Koekman A, et al. Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J Allergy Clin Immunol. 2016;138(5):1414-9.
47. Ivanov I, Matafonov A, Sun MF, Mohammed BM, Cheng Q, Dickeson SK, et al. A mechanism for hereditary angioedema with normal C1 inhibitor: an inhibitory regulatory role for the factor XII heavy chain. Blood. 2019;133(10):1152-63.
48. Bork K, Wulff K, Witzke G, Hardt J. Hereditary angioedema with normal C1-INH with versus without specific F12 gene mutations. Allergy. 2015;70(8):1004-12.
49. Bork K, Wulff K, Steinmüller-Magin L, Braenne I, Staubach-Renz P, Witzke G, et al. Hereditary angioedema with a mutation in the plasminogen gene. Allergy. 2018;73(2):442-50.
50. Bafunno V, Firinu D, D'Apolito M, Cordisco G, Loffredo S, Leccese A, et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J Allergy Clin Immunol. 2018;141(3):1009-17.
51. Bork K, Wulff K, Rossmann H, Steinmüller-Magin L, Braenne I, Witzke G, et al. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy. 2019;74(12):2479-81.
52. Ariano A, D'Apolito M, Bova M, Bellanti F, Loffredo S, D'Andrea G, et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy. 2020;75(11):2989-92.
53. Bork K, Wulff K, Möhl BS, Steinmüller-Magin L, Witzke G, Hardt J, et al. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J Allergy Clin Immunol. 2021;148(4):1041-8.
54. Bork K. Hereditary angioedema with normal C1 inhibitor. Immunol Allergy Clin North Am. 2013;33(4):457-70.
55. Saule C, Boccon-Gibod I, Fain O, Kanny G, Plu-Bureau G, Martin L, et al. Benefits of progestin contraception in non-allergic angioedema. Clin Exp Allergy. 2013;43(4):475-82.
56. Craig TJ, Bernstein JA, Farkas H, Bouillet L, Boccon-Gibod I. Diagnosis and treatment of bradykinin-mediated angioedema: outcomes from an angioedema expert consensus meeting. Int Arch Allergy Immunol. 2014;165(2):119-27.
57. Joseph K, Tholanikunnel BG, Kaplan AP. Cytokine and estrogen stimulation of endothelial cells augments activation of the prekallikrein-high molecular weight kininogen complex: Implications for hereditary angioedema. J Allergy Clin Immunol. 2017;140(1):170-6.
58. Citarella F, Misiti S, Felici A, Aiuti A, La Porta C, Fantoni A. The 5' sequence of human factor XII gene contains transcription regulatory elements typical of liver specific, estrogen-modulated genes. Biochim Biophys Acta. 1993;1172(1-2):197-9.
59. Gompel A, Fain O, Boccon-Gibod I, Gobert D, Bouillet L. Exogenous hormones and hereditary angioedema. Int Immunopharmacol. 2020;78:106080.
60. Banerji A, Davis KH, Brown TM, Hollis K, Hunter SM, Long J, et al. Patient-reported burden of hereditary angioedema: findings from a patient survey in the United States. Ann Allergy Asthma Immunol. 2020;124(6):600-7.
61. Grumach AS, Valle SO, Toledo E, de Moraes Vasconcelos D, Villela MM, Mansour E, et al; group interested on HAE (GINHA). Hereditary angioedema: first report of the Brazilian registry and challenges. J Eur Acad Dermatol Venereol. 2013;27(3):e338-44.
62. Lang DM, Aberer W, Bernstein JA, Chng HH, Grumach AS, Hide M, et al. International consensus on hereditary and acquired angioedema. Ann Allergy Asthma Immunol. 2012;109(6):395-402.
63. Schöffl C, Wiednig M, Koch L. Hereditary angioedema in Austria: prevalence and regional peculiarities. J Dtsch Dermatol Ges. 2019; 17(4):416-23.
64. Zanichelli A, Magerl M, Longhurst H, Fabien V, Maurer M. Hereditary angioedema with C1 inhibitor deficiency: delay in diagnosis in Europe. Allergy Asthma Clin Immunol. 2013;9(1):29.
65. Christiansen SC, Davis DK, Castaldo AJ, Zuraw BL. Pediatric hereditary angioedema: onset, diagnostic delay, and disease severity. Clin Pediatr (Phila). 2016;55(10):935-42.
66. Veronez CL, Moreno AS, Constantino-Silva RN, Maia LSM, Ferriani MPL, Castro FFM, et al. Hereditary Angioedema with Normal C1 Inhibitor and F12 Mutations in 42 Brazilian Families. J Allergy Clin Immunol Pract. 2018;6(4):1209-16.
67. Zuraw BL, Bork K, Binkley KE, Banerji A, Christiansen SC, Castaldo A, et al. Hereditary angioedema with normal C1 inhibitor function: consensus of an international expert panel. Allergy Asthma Proc. 2012;33 Suppl 1:S145-56.
68. Agostoni A, Cicardi M. Hereditary and acquired C1-inhibitor deficiency: biological and clinical characteristics in 235 patients. Medicine (Baltimore). 1992(4);71:206-15.
69. Bork K, Meng G, Staubach P, Hardt J. Hereditary angioedema: new findings concerning symptoms, affected organs, and course. Am J Med. 2006;119(3):267-74.
70. Cicardi M, Bergamaschini L, Marasini B, Boccassini G, Tucci A, Agostoni A. Hereditary angioedema: an appraisal of 104 cases. Am J Med Sci. 1982;284(1):2-9.
71. Betschel S, Badiou J, Binkley K, Borici-Mazi R, Hébert J, Kanani A, et al. The International/Canadian Hereditary Angioedema Guideline. Allergy Asthma Clin Immunol. 2019;15:72.
72. Busse PJ, Christiansen SC, Riedl MA, Banerji A, Bernstein JA, Castaldo AJ, et al. US HAEA Medical Advisory Board 2020 Guidelines for the Management of Hereditary Angioedema. J Allergy Clin Immunol Pract. 2021 Jan;9(1):132-50.
73. Bork K, Siedlecki K, Bosch S, Schopf RE, Kreuz W. Asphyxiation by laryngeal edema in patients with hereditary angioedema. Mayo Clin Proc. 2000;75(4):349-54.
74. Craig T, Aygören-Pürsün E, Bork K, Bowen T, Boysen H, Farkas H, et al. WAO guideline for the management of hereditary angioedema. World Allergy Organ J. 2012;5(12):182-99.
75. Longhurst HJ, Farkas H, Craig T, Aygören-Pürsün E, Bethune C, Bjorkander J, et al. HAE international home therapy consensus document. Allergy Asthma Clin Immunol. 2010;6(1):22.
76. Porebski G, Kwitniewski M, Reshef A. Biomarkers in Hereditary Angioedema. Clin Rev Allergy Immunol. 2021;60(3):404-13.
77. Farkas H, Martinez-Saguer I, Bork K, Bowen T, Craig T, Frank M, et al. International consensus on the diagnosis and management of pediatric patients with hereditary angioedema with C1 inhibitor deficiency. Allergy. 2017;72(2):300-13.
78. Agostoni A, Aygören-Pürsün E, Binkley KE, Blanch A, Bork K, Bouillet L, et al. Hereditary and acquired angioedema: problems and progress: proceedings of the third C1 esterase inhibitor deficiency workshop and beyond. J Allergy Clin Immunol. 2004;114(3 Suppl):S51-131.
79. Busse PJ, Christiansen SC. Hereditary Angioedema. N Engl J Med. 2020;382(12):1136-48.
80. Zotter Z, Csuka D, Szabo E, Czaller I, Nebenfuhrer Z, Temesszentandrasi G, et al. The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. 2014;9:44.
81. Caballero T, Maurer M, Longhurst HJ, Aberer W, Bouillet L, Fabien V, IOS Study Group. Triggers and prodromal symptoms of angioedema attacks in patients with hereditary angioedema. J Investig Allergol Clin Immunol. 2016(6);26:383-6.
82. Zuraw BL, Christiansen SC. HAE: pathophysiology and underlying mechanisms. Clin Rev Allergy Immunol. 2016;51(2):216-29.
83. Bork K, Hardt J, Staubach-Renz P, Witzke G. Risk of laryngeal edema and facial swellings after tooth extraction in patients with hereditary angioedema with and without prophylaxis with C1 inhibitor concentrate: a retrospective study. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2011;112(1):58-64.
84. Aygören-Pürsün E, Martinez Saguer I, Kreuz W, Klingebiel T, Schwabe D. Risk of angioedema following invasive or surgical procedures in HAE type I and II - the natural history. Allergy. 2013;68(8):1034-39.
85. Farkas H. Pediatric hereditary angioedema due to C1-inhibitor deficiency. Allergy Asthma Clin Immunol. 2010;6(1):8.
86. Caballero T, Farkas H, Bouillet L, Bowen T, Gompel A, Fagerberg C, et al.; C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency. J Allergy Clin Immunol. 2012; 129(2):308-20.
87. Hudey SN, Westermann-Clark E, Lockey RF. Cardiovascular and diabetic medications that cause bradykinin-mediated angioedema. J Allergy Clin Immunol Pract. 2017;5(3) 610-15.
88. Scott SI, Andersen MF, Aagaard L, Buchwald CV, Rasmussen ER. Dipeptidyl Peptidase-4 Inhibitor Induced Angioedema - An Overlooked Adverse Drug Reaction? Curr Diabetes Rev. 2018;14(4):327-33.
89. Andersen LK, Jensen JE, Bygum A. Second episode of near-fatal angioedema in a patient treated with everolimus. Ann Allergy Asthma Immunol. 2015;115(2):152-3.
90. Grumach AS, Staubach-Renz P, Villa RC, Diez-Zuluaga S, Reese I, Lumry WR. Triggers of exacerbation in chronic urticaria and recurrent angioedema-prevalence and relevance. J Allergy Clin Immunol Pract. 2021;9(6):2160-8.
91. Johnson FA, Wirth M, Zhu Z, Hahn J, Greve J, Ebert E, et al. Etiology and predictors of cluster attacks of hereditary angioedema that recur despite pharmaceutical treatment. Allergy Asthma Proc. 2021;42(4):317-24.
92. Kemp JG, Craig TJ. Variability of prodromal signs and symptoms associated with hereditary angioedema attacks: a literature review. Allergy Asthma Proc. 2009;30(5):493-9.
93. Leibovich-Nassi I, Reshef A. The Enigma of Prodromes in Hereditary Angioedema (HAE). Clin Rev Allergy Immunol. 2021;61(1):15-28.
94. Magerl M, Doumoulakis G, Kalkounou I, Weller K, Church MK, Kreuz W, et al. Characterization of prodromal symptoms in a large population of patients with hereditary angio-oedema. Clin Exp Dermatol. 2014;39(3):298-303.
95. Martinez-Saguer I, Farkas H. Erythema marginatum as an early symptom of hereditary angioedema: case report of 2 newborns. Pediatrics. 2016;137(2):e20152411.
96. Bork K, Gül D, Hardt J, Dewald G. Hereditary angioedema with normal C1 inhibitor: clinical symptoms and course. Am J Med. 2007;120(11):987-92.
97. Firinu D, Bafunno V, Vecchione G, Barca MP, Manconi PE, Santacroce R, et al. Characterization of patients with angioedema without wheals: the importance of F12 gene screening. Clin Immunol. 2015;157(2):239-48.
98. Piñero-Saavedra M, González-Quevedo T, Saenz de San Pedro B, Alcaraz C, Bobadilla-González P, Fernández-Vieira L, et al. Hereditary angioedema with F12 mutation: Clinical features and enzyme polymorphisms in 9 Southwestern Spanish families. Ann Allergy Asthma Immunol. 2016;117(5):520-6.
99. Taya J, Veronez CL, Pesquero JB, Bork K, Grumach AS. Uncommon signs associated with hereditary angioedema with normal C1 inhibitor. J Investig Allergol Clin Immunol. 2021;31(3):257-8.
100. Rasmussen ER, de Freitas PV, Bygum A. Urticaria and prodromal symptoms including erythema marginatum in Danish patients with hereditary angioedema. Acta Derm Venereol. 2016;96(3):373-6.
101. Gutierrez M, Veronez CL, Rodrigues Valle SO, Gonçalves RF, Ferriani MPL, Moreno AS, et al. Unnecessary abdominal surgeries in attacks of hereditary angioedema with normal C1 inhibitor. Clin Rev Allergy Immunol. 2021;61(1):60-5.
102. Mormile I, Cocchiaro A, Bova M, Loffredo S, de Paulis A, Spadaro G, et al. Gastrointestinal manifestations of angioedema: a potential area of misdiagnosis. Eur J Gastroenterol Hepatol. 2021;33(6):787-93.
103. Gábos G, Dobru D, Mihály E, Bara N, Dumitrache C, Popa R, et al. Recurrent ascites: a need to evaluate for hereditary angio-oedema. Lancet. 2017;390(10107):2119-20.
104. Bork K, Hardt J, Schicketanz KH, Ressel N. Clinical studies of sudden upper airway obstruction in patients with hereditary angioedema due to C1 esterase inhibitor deficiency Arch Intern Med. 2003;163(10):1229-35.
105. Bork K, Hardt J, Witzke G. Fatal laryngeal attacks and mortality in hereditary angioedema due to C1-INH deficiency. J Allergy Clin Immunol. 2012;130(3):692-7.
106. Zanichelli A, Arcoleo F, Barca MP, Borrelli P, Bova M, Cancian M, et al. A nationwide survey of hereditary angioedema due to C1 inhibitor deficiency in Italy. Orphanet J Rare Dis. 2015;10:11.
107. Lin CT, Shyur SD, Fang LC, Huang HH, Shih YY. Unusual presentation of linear wrist blisters associated with hereditary angioedema: The first case report in Taiwan. J Formos Med Assoc. 2021;120(8):1642-6.
108. Serpa FS, Veronez CL, Campinhos FL, Moyses TR, Pesquero JB. SERPING1 mutation in a rare hereditary angioedema with skin blisters. Ann Allergy Asthma Immunol. 2019;122(3):340-1.
109. Veronez CL, Campos RA, Constantino-Silva RN, Nicolicht P, Pesquero JB, Grumach AS. Hereditary Angioedema-Associated Acute Pancreatitis in C1-Inhibitor Deficient and Normal C1-Inhibitor Patients: Case Reports and Literature Review. Front Med (Lausanne). 2019;6:80.
110. Tarzi MD, Hickey A, Förster T, Mohammadi M, Longhurst HJ. An evaluation of tests used for the diagnosis and monitoring of C1 inhibitor deficiency: normal serum C4 does not exclude hereditary angio-oedema. Clin Exp Immunol. 2007;149(3):513-6.
111. Veronez CL, Grumach AS. Angioedema without urticaria: novel findings which must be measured in clinical setting. Curr Opin Allergy Clin Immunol. 2020;20(3):253-60.
112. Gompels MM, Lock RJ, Unsworth DJ, Johnston SL, Archer CB, Davies SV. Misdiagnosis of hereditary angio-oedema type 1 and type 2. Br J Dermatol. 2003;148(4):719-23.
113. Honda D, Ohsawa I, Mano S, Rinno H, Tomino Y, Suzuki Y. Cut-off value of C1-inhibitor function for the diagnosis of hereditary angioedema due to C1-inhibitor deficiency. Intractable Rare Dis Res. 2021;10(1):42-7.
114. Bork K, Staubach-Renz P, Hardt J. Angioedema due to acquired C1-inhibitor deficiency: spectrum and treatment with C1-inhibitor concentrate. Orphanet J Rare Dis. 2019;14(1):65.
115. Zanichelli A, Azin GM, Wu MA, Suffritti C, Maggioni L, Caccia S, et al. Diagnosis, course, and management of angioedema in patients with acquired C1-inhibitor deficiency. J Allergy Clin Immunol Pract. 2017;5(5):1307-13.
116. Germenis AE, Rijavec M, Veronez CL. Leveraging genetics for Hereditary Angioedema: A road map to precision medicine. Clin Rev Allergy Immunol. 2021;60(3):416-28.
117. Veronez CL, Mendes AR, Leite CS, Gomes CP, Grumach AS, Pesquero JB; Hereditary Angioedema Brazilian Study Group (GEBRAEH). The panorama of primary angioedema in the Brazilian population. J Allergy Clin Immunol Pract. 2021;9(6):2293-304.
118. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24.
119. Belbézier A, Bocquet A, Bouillet L. Idiopathic Angioedema: current challenges. J Asthma Allergy. 2020;13:137-44.
120. Iriarte Sotés P, Armisén M, Usero-Bárcena T, Rodriguez Fernández A, Otero Rivas MM, Gonzalez MT, et al. Efficacy and safety of up-dosing antihistamines in chronic spontaneous urticaria: a systematic review of the literature. J Investig Allergol Clin Immunol. 2021;31(4):282-91.
121. Crochet J, Lepelley M, Yahiaoui N, Vermorel C, Bosson JL, Pralong P, et al. Bradykinin mechanism is the main responsible for death by isolated asphyxiating angioedema in France. Clin Exp Allergy. 2019;49(2):252-4.
122. Beltrami L, Zanichelli A, Zingale L, Vacchini R, Carugo S, Cicardi M. Long-term follow-up of 111 patients with angiotensin converting enzyme inhibitor-related angioedema. J Hypertens. 2011;29(11):2273-7.
123. Makani H, Messerli FH, Romero J, Wever-Pinzon O, Korniyenko A, Berrios RS, et al. Meta-analysis of randomized trials of angioedema as an adverse event of renin-angiotensin system inhibitors. Am J Cardiol. 2012;110(3):383-91.
124. Ferriani MPL, Trevisan-Neto O, Costa JS, Melo JML, Moreno AS, Dias MM, et al. Acquired angioedema due to C1 inhibitor deficiency preceding splenic marginal zone lymphoma: further insights from clinical practice. Int Arch Allergy Immunol. 2020;181(12):941-6.
125. Zingale LC, Castelli R, Zanichelli A, Cicardi M. Acquired deficiency of the inhibitor of the first complement component: presentation, diagnosis, course, and conventional management. Immunol Allergy Clin North Am. 2006(4);26:669-90.
126. Hofman ZLM, Van den Elzen MT, Kuijpers J, de Maat S, Hack CE, Knulst AC, et al. Evidence for bradykinin release in chronic spontaneous urticaria. Clin Exp Allergy. 2020;50(3):343-51.
127. Oschatz C, Maas C, Lecher B, Jansen T, Björkqvist J, Tradler T, et al. Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity. 2011;34(2):258-68.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888