Número Atual: Janeiro-Março 2022 - Volume 6 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores

Comunicações Clínicas e Experimentais
Anemia hemolítica autoimune na doença de Castleman multicêntrica: relato de caso
Autoimmune hemolytic anemia in multicentric Castleman's disease: case report
Marcos Tadeu Nolasco da-Silva1; Katariny Parreira de Oliveira Alves2; Izilda Aparecida Cardinalli2; Amanda Avesani Cavotto Furlan3; Priscila Machado Fernandes1
DOI: 10.5935/2526-5393.20220013
1. Universidade Estadual de Campinas, Departamento de Pediatria - Campinas, SP, Brasil
2. Universidade Estadual de Campinas, Departamento de Anatomia Patológica - Campinas, SP, Brasil
3. Universidade Estadual de Campinas, Departamento de Clínica Médica - Campinas, SP, Brasil
Endereço para correspondência:
Amanda Avesani Cavotto Furlan
E-mail: furlan.amanda@gmail.com
Submetido em: 18/10/2021
Aceito em: 13/11/2021
Não foram declarados conflitos de interesse associados à publicação deste artigo.
RESUMO
A doença de Castleman é um distúrbio linfoproliferativo raro, podendo se manifestar sob a forma de massas localizadas ou como doença multicêntrica. A doença de Castleman multicêntrica é caracterizada por adenopatias generalizadas, visceromegalias, manifestações autoimunes e infecções recorrentes. Este artigo apresenta o relato de caso de anemia hemolítica autoimune por anticorpos quentes em paciente com doença de Castleman multicêntrica. Resposta eficaz foi obtida com uso de corticoterapia sistêmica e tocilizumabe.
Descritores: Hiperplasia do linfonodo gigante; anemia hemolítica autoimune; anticorpos monoclonais.
INTRODUÇÃO
Descrita pela primeira vez em 1954 por Castleman e Towne1, a doença de Castleman (DC) é um distúrbio linfoproliferativo raro policlonal de linfócitos B e plasmócitos, podendo se manifestar sob a forma de doença unicêntrica ou multicêntrica2.
HISTOPATOLOGIA
A DC pode ser subdividida em duas principais formas histopatológicas: variante hialino-vascular, e variante de células plasmáticas3. A variante mista apresenta elementos de ambas as variantes, e está presente em aproximadamente 10% dos casos. Há ainda a subvariante plasmablástica, associada à infecção por HHV-8 (herpes vírus humano tipo 8) e HIV (vírus da imunodeficiência humana), com risco de progressão para linfoma monoclonal plasmablástico.
Histologicamente, a variante hialino-vascular clássica se caracteriza por distorção da arquitetura linfonodal. Observa-se aumento do número de folículos linfoides, com variação de tamanho e forma. Uma das lesões identificadas é a atresia folicular, com linfócitos da zona do manto dispostos em camadas em torno do centro folicular (aspecto em "casca de cebola"). Nota-se deposição de material hialino nos centros germinativos, destacado pela reação do ácido periódico de Schiff (PAS). Alterações vasculares, como esclerose dos vasos que penetram os folículos, originam lesões conhecidas como lollipop follicles4.
As características histopatológicas da variante de células plasmáticas mostram diferenças nas formas HHV-8 negativo e HHV-8 positivo. Os casos HHV-8 negativo apresentam aumento no número de plasmócitos maduros agregados, principalmente, nas áreas interfoliculares. A proliferação vascular na região paracortical é proeminente. Os folículos se encontram hiperplásicos ou normais. Nas formas HHV-8 positivas há expansão da zona interfolicular por plasmócitos imaturos e maduros, com variável grau de atipia. Além disso, observa-se menor distinção do limite entre a zona do manto e a região interfolicular. A variante mista, histologicamente, apresenta uma sobreposição das duas variantes; hialino-vascular e de células plasmáticas5.
A doença de Castleman unicêntrica (DCU) está comumente associada à variante hialino-vascular, e correspondente a aproximadamente 70% dos casos. É caracterizada por linfonodomegalias localizadas, geralmente em mediastino ou abdome, em pacientes oligo ou assintomáticos, em que a ressecção do linfonodo acometido resulta na cura da doença6.
A doença de Castleman multicêntrica (DCM) está comumente associada à variante de células plasmáticas, e corresponde a aproximadamente 10-20% dos casos. É caracterizada por linfonodomegalias difusas e sintomas sistêmicos moderados a severos, dentre os quais: febre, sudorese noturna, fraqueza, anorexia e perda de peso. Características clínicas incluem também hepatoesplenomegalia, ascite, derrame pericárdico, derrame pleural e rash cutâneo. Anormalidades laboratoriais incluem anemia de doença crônica, trombocitopenia, hipoalbuminemia, hipergamaglobulinemia policlonal, aumento de VHS (velocidade de hemossedimentação), proteína C-reativa, IL-6 (interleucina 6) e VEGF (fator de crescimento endotelial vascular).
ETIOLOGIA
A DCM pode estar associada à infecção por HHV-8, com ou sem coinfecção com HIV, em até 50% dos casos. Mais raramente, também pode estar associada à síndrome POEMS (síndrome paraneoplásica caracterizada por polineuropatia, organomegalia, endocrinopatia, gamopatia monoclonal e alterações cutâneas). Na outra metade dos casos, a DCM é considerada idiopática7.
A etiologia da DCM idiopática é incerta, e suas teorias incluem: autoanticorpos patológicos ou mutações na regulação do sistema imune inato (hipótese autoimune/autoinflamatória); presença de pequena população de células neoplásicas (hipótese paraneoplásica); ou presença de algum outro vírus não identificado (hipótese viral)8. Foi descrita ainda uma variante severa da DCM idiopática conhecida por síndrome TAFRO, que contempla trombocitopenia, anasarca, febre, fibrose da reticulina e organomegália em seus aspectos clínicos9.
Sabe-se, também, que a superprodução de interleucinas pró-inflamatórias, em especial a IL-6, está implicada na fisiopatologia da doença.
A DCM também cursa com infecções recorrentes, incluindo oportunistas, e manifestações autoimunes. A ocorrência, entretanto, de anemia hemolítica autoimune, é rara, tendo sidos descritos até a presente data, de que os autores desse relato tenham conhecimento, seis casos10-15.
INTERLEUCINA-6 E TOCILIZUMABE
A IL-6 é uma citocina pró-inflamatória que induz a diferenciação e proliferação de linfócitos T e B, está envolvida na síntese de proteínas de fase aguda, no estímulo à hematopoiese e à produção hepática de hepcidina e no desenvolvimento de sintomas constitucionais presentes em várias doenças inflamatórias. Na DC, os linfonodos hiperplásicos com infiltração de plasmócitos apresentam produção constitucionalmente aumentada de IL-616.
Para confirmar o envolvimento da IL-6 na fisiopatologia da DC, em 198617 foi realizado um estudo com dois pacientes afetados pela DC (um na forma multicêntrica, e outro, unicêntrica). Ambos foram submetidos à ressecção da maior cadeia linfonodal acometida - no caso da DCU, a única acometida. A cultura do sobrenadante dos linfonodos ressecados confirmou a produção de IL-6. Observou-se também que o paciente com DCU apresentou remissão completa dos sintomas após o procedimento, enquanto que o paciente com DCM manteve níveis séricos elevados de IL-6 e persistências dos sintomas.
Yoshizaki e cols., em estudo de 1989, também demonstraram associação entre os níveis de IL-6, hiperplasia linfonodal, hipergamaglobulinemia, níveis de proteína C-reativa e anormalidade clínicas na DC18.
Desta forma, estudos se seguiram procurando terapias anti-IL-6 eficazes no tratamento da doença. A primeira droga a demonstrar resultados promissores foi desenvolvida no Japão, na forma de um anticorpo monoclonal antirreceptor de IL-6: o tocilizumabe. Inicialmente desenvolvido para o tratamento de artrite reumatoide no final da década de 90, o tocilizumabe foi também aprovado para estudos clínicos na DC e na artrite idiopática juvenil no início do século XXI.
Os primeiros estudos com a administração do tocilizumabe em pacientes com DCM ocorreram também no Japão19,20 e demonstraram reversão dos parâmetros inflamatórios, resolução dos sintomas constitucionais e redução dos níveis de linfadenopatia. Os resultados se mantiveram após três anos de uso contínuo da medicação, e permitiu também o desmame da corticoterapia sistêmica em parte dos pacientes que a haviam iniciado concomitantemente.
Desde então, o tocilizumabe vem se consagrado importante terapêutica a ser considerada nos pacientes acometidos pela DCM idiopática. Entretanto, tendo em vista sua etiologia incerta e o acometimento multisistêmico grave da doença, ainda não foi descoberta uma cura definitiva, e o prognóstico da doença permanece desfavorável.
RELATO DE CASO
Foi admitido em agosto de 2020 no Hospital de Clínicas da UNICAMP paciente masculino de 25 anos com quadro de cefaleia há 6 dias associada a quadro de dispneia aos esforços, vômitos esverdeados, inapetência e icterícia. Negava febre, sintomas respiratórios, alteração de hábito intestinal ou urinário, incluindo acolia fecal ou colúria, viagens recentes, picadas de insetos e ingestão de alimentos suspeitos ou medicações.
Paciente portador de doença de Castelman multicêntrica idiopática, obteve confirmação diagnóstica em 2006 por meio de biópsia de linfonodo mesentérico. Durante o curso pregresso da doença, manifestara-se com quadro inflamatório crônico - marcado por períodos recorrentes de febre, emagrecimento, adinamia e anorexia - infecções oportunistas recorrentes e intercorrências imunomediadas, como glomerulonefrite e miocardiopatia.
A seguir, os cortes histológicos do linfonodo mesentérico ressecado em 2006 mostram distorção da arquitetura linfonodal, com expansão da região paracortical por aumento do número de plasmócitos maduros (Figura 1), associada a proeminente vascularização (Figura 2). Identifica-se hiperplasia folicular, porém com centros germinativos regredidos, alguns com aspecto em "casca de cebola" (Figura 3).
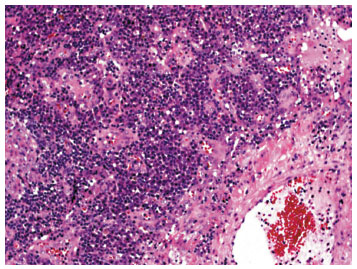
Figura 1
Evidente aumento no número de plasmócitos provocando expansão da zona paracortical linfonodal (H&E, 100x)
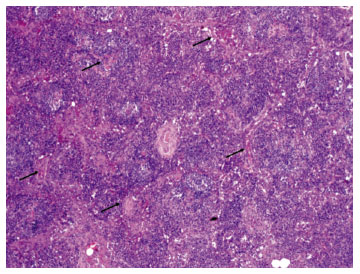
Figura 2
Proeminente vascularização interfolicular (setas). Notar a moderada distorção da arquitetura linfonodal (H&E, 40x)
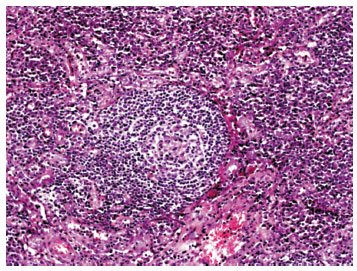
Figura 3
Regressão de folículo linfoide na zona cortical (H&E, 100x)
O estudo imuno-histoquímico mostra CD20 positivo em linfócitos B nos folículos regressivos, CD3 positivo em linfócitos T interfoliculares, kappa e lambda positivos em frequentes plasmócitos em padrão policlonal (Figura 4). Os marcadores CD30, HHV8 e a hibridização in situ resultaram negativos. Dessa forma, o estudo imuno-histoquímico corroborou o diagnóstico de doença de Castleman, variante histopatológica mista.
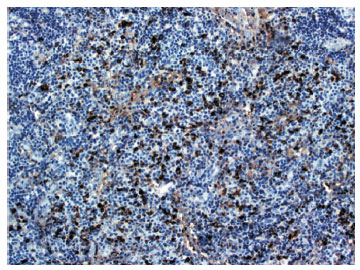
Figura 4
Proeminente aumento de plasmócitos na zona paracortical (reação imuno-histoquímica para cadeia leve lambda, 100x)
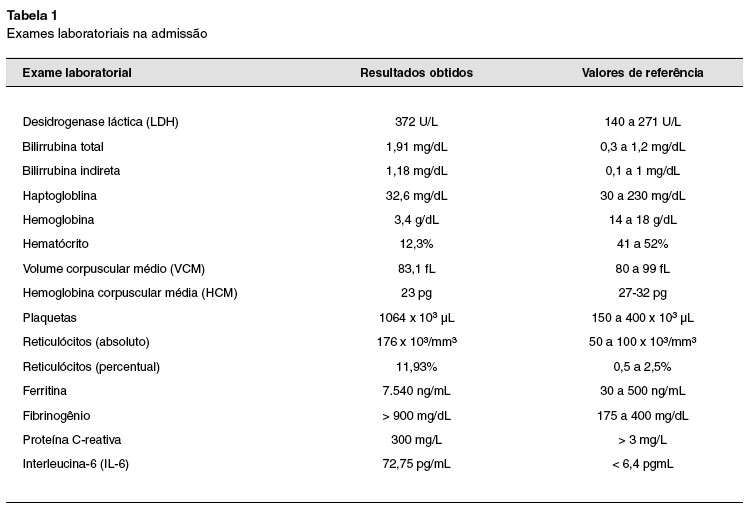
Ao exame físico atual, apresentava-se com palidez cutâneo-mucosa, icterícia leve e taquicardia leve, sem hepato ou esplenomegalia. Exames laboratoriais mostraram hemoglobina 3,4 g/dL, volume corpuscular médio (VCM) 83,1 fl, plaquetas 1.064.000 µL, haptoglobina 32,6 mg/dL , reticulócitos totais 176.000/mm3, LDH 372 U/L, bilirrubina indireta 1,18 mg/dL, teste de antiglobulina direto positivo (IgG 1+, C3d 3+) e eluato positivo, com presença de autoanticorpos classe IgG sem especificidade definida. No esfregaço de sangue periférico, não foram encontrados esquizócitos ou outras formas anômalas de hemácias.
Sorologias para hepatite A, hepatite B, hepatite C, sífilis e HIV resultaram negativas. Urina I negativa para hemoglobinúria. Ultrassom abdome total sem anormalidades. Ainda, triglicérides 156 mg/dL, ferritina 7540 ng/mL, fibrinogênio > 900 mg/dL e proteína C-reativa 300 mg/L. AST, ALT, amilase, lipase, crioaglutininas, albumina e coagulograma normais.
Os exames laboratoriais alterados da admissão estão representados na Tabela 1.
Com base nos dados acima, o diagnóstico de anemia hemolítica autoimune por anticorpos quentes foi definido. De início foi realizada transfusão de uma unidade de concentrado de hemácias, sem elevação significativa dos níveis de hemoglobina (3,4 g/dL para 4,3 g/dL). Após confirmação diagnóstica, o tratamento foi iniciado com corticoterapia sistêmica (metilprednisolona 4 mg/kg/dia) e, dois dias após, tocilizumabe 4 mg/kg dose única.
No primeiro dia após a administração do tocilizumabe, observou-se aumento progressivo dos níveis de hemoglobina, com normalização dos níveis no oitavo dia. Também foi observada normalização dos níveis de LDH, bilirrubina e haptoglobina. Iniciado desmame progressivo do corticoide a partir do sétimo dia.
A resposta ao tratamento administrado está representada nas Figuras 5 e 6.
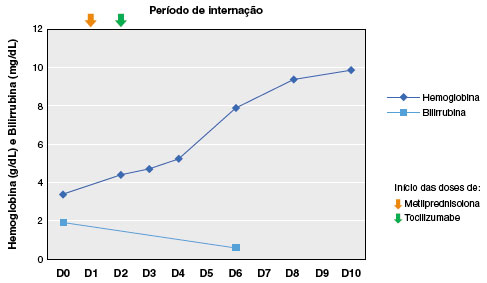
Figura 5
Resposta ao tratamento
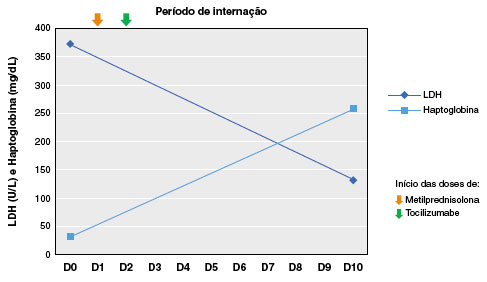
Figura 6
Resposta ao tratamento
O paciente manteve retornos regulares após a alta hospitalar, com administração mensal de tocilizumabe 8 mg/kg em monoterapia, com resposta sustentada até a presente data.
DISCUSSÃO
A existência de íntima relação entre autoimunidade e doenças linfoproliferativas é conhecida, e tem como base a fisiopatologia da proliferação, transformação e autorreatividade dos linfócitos B21. É inerente a qualquer linfócito B produzir anticorpos de baixa afinidade contra autoantígenos, que são eliminados pelos mecanismos imunorregulatórios assim que são reconhecidos. Porém, nas doenças linfoproliferativas, as mutações nas linhagens germinativas, a alta atividade proliferativa dos linfócitos B e o defeito no mecanismo de apoptose levam à desregulação do sistema imune e à geração de autoanticorpos.
As doenças linfoproliferativas que mais cursam com doenças autoimunes são: mieloma múltiplo, gamopatia monoclonal de significado indeterminado, linfoma não Hodgkin e leucemia linfoide crônica. Nesta última, a ocorrência de anemia hemolítica autoimune ocorre em até 20-25% dos pacientes no curso da doença22.
Doenças autoimunes já foram reportadas em pacientes com DCM idiopática, como lúpus eritematoso sistêmico e linfohistiocitose hemofagocítica23. Porém, raros são os relatos de anemia hemolítica autoimune.
Dentre os sinais e sintomas sistêmicos presentes na DCM, a anemia invarialmente está quase sempre presente, geralmente com as características típicas de anemia de doença crônica24. A patogênese da anemia de doença crônica nesses pacientes envolve os mesmos determinantes que outras doenças inflamatórias, com o mecanismo adicional da IL-6, que possui atividade inibitória sobre a eritropoese. Entretanto, é possível que a própria superprodução de IL-6 possa estar envolvida na fisiopatologia da anemia hemolítica autoimune (AHAI) através do estímulo à geração e diferenciação de plasmócitos.
Há mais de 20 anos, modelos experimentais de indução de autoimunidade, como a artrite induzida por colágeno e a artrite induzida por antígeno, utilizam-se da IL-625. Também em pacientes com DCM e AHAI, estudos mostram níveis aumentados de IL-6, assim como de IL-4, IL-10, IL-13, IL-17 e IL-2126. Além disso, a presença de células T helper 2 (Th2), T reguladoras (Treg) e T helper 17 (Th17) sugerem sua associação com a atividade da doença27. Células Th2 secretam IL-6, IL-4, IL-10, IL-13 e TGF-b, citocinas que estimulam a produção de anticorpos pelos linfócitos B, enquanto que a IL-6 induz a diferenciação das células Th17, ampliando a resposta proinflamatória e autoimune28,29.
Até o momento, somente um estudo relatou sucesso no tratamento da AHAI na DC com a utilização do tocilizumabe30, enquanto que estudo mais recente de 2019 demonstrou sucesso parcial. Neste estudo, Tabata S. e cols. relatam o caso de um paciente com DCM idiopática com AHAI, sendo estabelecido tratamento inicial com tocilizumabe 8 mg/kg a cada duas semanas no total de 6 doses. Porém, após a segunda dose da medicação, houve a detecção de anticorpos anti-tocilizumabe no soro do paciente, com consequente diminuição da eficácia do tratamento. No entanto, observa-se que houve introdução tardia da corticoterapia em dose imunossupressora nesse paciente (seis dias após o início do tocilizumabe).
Nosso estudo permitiu a utilização do tocilizumabe na dose 4 mg/kg, à medida que associou a metilprednisolona na dose 4 mg/kg também. Essa associação permitiu não só reduzir a dose de tocilizumabe, o que torna o custo do tratamento mais acessível, como também evitou o possível surgimento de anticorpos contra a droga. Entretanto, cabe ressaltar que a alta dose de corticoterapia, mesmo que seguida de desmame completo, trouxe como efeito colateral ao paciente deste estudo acne moderada e descontrole glicêmico. É possível supor que a dose de 1-2 mg/kg também teria efeito imunossupressor, porém seria acompanhada de menos efeitos colaterais.
O tocilizumabe foi aprovado no Japão para o tratamento da DCM idiopática em 2005, porém devido à falta de estudos randomizados controlados, não foi aprovado nos Estados Unidos, onde em 2014 foi aprovada outra droga semelhante, denominada siltuximabe, anticorpo monoclonal anti IL-6. Embora considerada hoje tratamento de escolha, apenas 34% dos pacientes responderam à terapia com o siltuximabe. Metade dos pacientes que não responderam apresentava baixa dosagem sérica de IL-631.
As vias de sinalização implicadas na patogênese da DCM idiopática ainda não estão totalmente esclarecidas, e outras terapias envolvendo vias consequentes à ativação da IL-6 também têm sido estudadas.
CONCLUSÃO
Novas opções terapêuticas para a DCM idiopática têm surgido e vêm demonstrando resultados promissores. Por ser doença rara, de etiologia incerta e com extensa variedade de manifestações clínicas, a DCM idiopática ainda consiste em grande desafio para a comunidade científica e permanece alvo de estudo. Entretanto, sabe-se que a IL-6 exerce papel fundamental na fisiopatologia da doença, e a inclusão dos anticorpos monoclonais antirreceptor IL-6 e anti-IL-6 no rol de tratamento mudou radicalmente o prognóstico destes pacientes.
REFERÊNCIAS
1. Castleman B, Towne VW. Case records of the Massachusetts General Hospital: Case No. 40231. N Engl J Med. 1954;250(23):1001-5.
2. Izuchukwu IS, Tourbaf K, Mahoney MC. An unusual presentation of Castleman's disease: a case report. BMC Infect Dis. 2003;3:20.
3. Beck JT, Hsu SM, Wijdenes J, Bataille R, Klein B, Vesole D, et al. Brief report: alleviation of systemic manifestations of Castleman's disease by monoclonal anti-interleukin-6 antibody. N Engl J Med. 1994;330(9):602-5.
4. Wu S, Lim MS, Jaffe ES. Pathology of Castleman Disease. Hematol Oncol Clin N Am. 2018;32:37-52.
5. Ioachim HL, Medeiros LJ. Ioachim´s lymph node pathology. Philadelphia: Wolters Kluwer/Lippincott Williams&Wilkins; 2009. p. 228-37. ISBN/ISSN: 9780781775960.
6. Saeed-Abdul-Rahman I, Al-Amri AM. Castleman disease. Korean J Hematol. 2012;47(3):163-77.
7. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646-57.
8. Fajgenbaum DC, van Rhee F, Nabel CS. HHV-8-negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123:2924-33.
9. Fajgenbaum DC, Uldrick TS, Bagg A, Frank D, Wu D, Srkalovic G, et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646-57.
10. Liberato NL, Bollati P, Chiofalo F, Filipponi M, Poli M. Autoimmune hemolytic anemia in multicentric Castleman's disease. Haematologica. 1996;81:40-3.
11. Ocio EM, Sanchez-Guijo FM, Diez-Campelo M, Castilla C, Blanco OJ, Caballero D, et al. Efficacy of rituximab in an aggressive form of multicentric Castleman disease associated with immune phenomena. Am J Hematol. 2005;78(4):302-5.
12. Tajima K, Yamamoto H, Suzuki I, Kato Y, Hatano K, Takahashi S, et al. Autoimmune hemolytic anemia with warm-reactive immunoglobulin M antibody in multicentric Castleman disease. Ann Hematol. 2013;92(6):849-51.
13. Yuzuriha A, Saitoh T, Koiso H, Mitsui T, Uchiumi H, Yokohama A, et al. Successful treatment of autoimmune hemolytic anemia associated with multicentric Castleman disease by anti-interleukin-6 receptor antibody (tocilizumab) therapy. Acta Haematol. 2011;126(3):147-50.
14. Hisatake J, Ishiyama T, Akimoto Y, Matsuda I, Hino K, Tomoyasu S, et al. [Autoimmune hemolytic anemia associated with multicentric Castleman's disease with a 28-year history]. Rinsho Ketsueki. 1994;35(8):768-73.
15. Tabata S, Higuchi T, Tatsukawa S, Narimatsu K, Takeo H, Matsukuma S, et al. Idiopathic Multicentric Castleman Disease with Autoimmune Hemolytic Anemia and Production of Anti-drug Antibody against Tocilizumab. Intern Med. 2019;58(22):3313-8.
16. Yoshizaki K, Murayama S, Ito H, Koga T. The Role of Interleukin-6 in Castleman Disease. Hematol Oncol Clin North Am. 2018;32(1):23-36.
17. Yoshizaki K, Matsuda T, Nishimoto N, Kuritani T, Taeho L, Aozasa K, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood. 1989;74(4):1360-7.
18. Yoshizaki K, Matsuda T, Nishimoto N, Kuritani T, Taeho L, Aozasa K, et al. Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman's disease. Blood. 1989;74(4):1360-7. PMID: 2788466.
19. Nishimoto N, Sasai M, Shima Y, Nakagawa M, Matsumoto T, Shirai T, et al. Improvement in Castleman's disease by humanized anti-interleukin-6 receptor antibody therapy. Blood. 2000;95(1):56-61.
20. Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627-32.
21. Caligaris-Cappio F, Bertero MT, Bergui L. Autoimmunity, autoimmune diseases and lymphoproliferative disorders. Haematologica. 1994;79(6):487-92.
22. Kipps TJ, Carson DA. Autoantibodies in chronic lymphocytic leukemia and related systemic autoimmune diseases. Blood. 1993;81:2475-87.
23. Liu AY, Nabel CS, Finkelman BS, Ruth JR, Kurzrock R, van Rhee F, et al. Idiopathic multicentric Castleman's disease: a systematic literature review. Lancet Haematol. 2016;3(4):e163-75.
24. Featherstone T, Bayliss AP, Ewen SWB, Brunt PW, Dawson AA. Obscure anemia and hepatic dysfunction in Castleman's disease. Gut. 1990;31:834-7.
25. Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, et al. Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med. 1998;187(4):461-8.
26. van Rhee F, Voorhees P, Dispenzieri A, Fosså A, Srkalovic G, Ide M, et al. International, evidence-based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115-24.
27. Kamesaki T. [Recent progress of diagnosis and treatment for immune-mediated hematological diseases. Topics: III. Diagnosis and treatment; 2. Autoimmune hemolytic anemia]. Nihon Naika Gakkai Zasshi. 2014;103(7):1599-608.
28. Xu L, Zhang T, Liu Z, Li Q, Xu Z, Ren T. Critical role of Th17 cells in development of autoimmune hemolytic anemia. Exp Hematol. 2012;40:994-1004.
29. Kimura A, Kishimoto T. IL-6: Regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830-5.
30. Kunitomi A, Konaka Y, Yagita M, Nishimoto N, Kishimoto T, Takatsuki K. Humanized anti-interleukin 6 receptor antibody induced long-term remission in a patient with life-threatening refractory autoimmune hemolytic anemia. Int J Hematol. 2004;80:246-9.
31. Casper C, Chaturvedi S, Munshi N, Wong R, Qi M, Schaffer M, et al. Analysis of Inflammatory and Anemia-Related Biomarkers in a Randomized, Double-Blind, Placebo-Controlled Study of Siltuximab (Anti-IL6 Monoclonal Antibody) in Patients With Multicentric Castleman Disease. Clin Cancer Res. 2015;21(19):4294-304.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888