Número Atual: Janeiro-Março 2021 - Volume 5 - Número 1
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Simone Cristina Soares Brandão
- Emmanuelle Tenório Albuquerque Madruga Godoi
- Lúcia Helena de Oliveira Cordeiro
- Débora Nóbrega de Lima
- Camila de Holanda Medeiros
- João Paulo Vieira e Silva de Albuquerque
- Maria Eduarda Lins Arraes Ramos
- Romero Carvalho Coimbra Albêlo
- Maria Fernanda Peixoto Matos
- Emanuel Sávio Cavalcanti Sarinho

Artigo de Revisão
Papel do imunometabolismo, receptores Toll-Like e ECA 2 na COVID-19
Role of immunometabolism, Toll-Like receptors, and ACE-2 in COVID-19
Simone Cristina Soares Brandão1-2; Emmanuelle Tenório Albuquerque Madruga Godoi1,2; Lúcia Helena de Oliveira Cordeiro1; Débora Nóbrega de Lima3; Camila de Holanda Medeiros3; João Paulo Vieira e Silva de Albuquerque4; Maria Eduarda Lins Arraes Ramos4; Romero Carvalho Coimbra Albêlo3; Maria Fernanda Peixoto Matos4; Emanuel Sávio Cavalcanti Sarinho5-6
1. Departamento de Medicina Interna da Universidade Federal de Pernambuco (UFPE), Recife, PE, Brasil
2. Pós-Graduação do Programa de Cirurgia, Universidade Federal de Pernambuco (UFPE), Recife, PE, Brasil
3. Graduandos da Universidade Federal de Pernambuco (UFPE), Recife, PE, Brasil
4. Graduandos da Faculdade Pernambucana de Saúde (FPS), Recife, PE, Brasil
5. Professor Titular da Universidade Federal de Pernambuco (UFPE)
6. Pós-Graduação em Saúde Translacional e da Pós-Graduação em Saúde da Criança e do Adolescente da Universidade Federal de Pernambuco (UFPE)
Endereço para correspondência:
Emanuel Sávio Cavalcanti Sarinho
E-mail: emanuel.sarinho@ufpe.br
Submetido em: 15/03/2021
Aceito em: 25/03/2021
RESUMO
No combate à infecção pelo coronavírus 2 da síndrome respiratória aguda grave (SARS-CoV-2), o organismo se utiliza de mecanismos da imunidade inata, dentre eles os receptores Toll- Like (TLR), responsáveis pela sinalização da inflamação através da liberação de mediadores químicos e recrutamento de células imunitárias. Na patologia causada pela doença do SARS-CoV-2 2019 (COVID-19), ganha especial importância o TLR-4, visto que a sua estimulação exacerbada vem sendo relacionada ao estado hiperinflamatório em fases avançadas da COVID-19. Outro receptor que desempenha um papel primordial na infecção pelo SARS-CoV-2, servindo como porta de entrada para o vírus e progressão da doença, é a enzima conversora de angiotensina 2 (ECA 2), cuja ligação com a proteína S viral causa desregulação de vários sistemas fundamentais para a homeostase, como o sistema renina-angiotensina-aldosterona. Pacientes com doenças cardiometabólicas como obesidade, diabetes, aterosclerose e hipertensão vêm sendo classificados como alto risco para desenvolver as formas graves da COVID-19, visto que o estado inflamatório, já existente nessas doenças, pode ser agravado pelo desequilíbrio metabólico causado pelo SARS-CoV-2. A elucidação desses e de outros mecanismos relacionados à fisiopatologia da COVID-19 é imprescindível para uma melhora na estratificação de risco, nas escolhas terapêuticas e no prognóstico desses pacientes. Desta forma, nesta revisão objetivamos discutir as relações entre TLR-4, ECA 2, doenças cardiometabólicas, infecção pelo SARS-CoV-2 e gravidade da COVID-19.
Descritores: SARS-CoV-2, COVID-19, receptor Toll-Like 4, enzima conversora de angiotensina 2, obesidade, aterosclerose, hipertensão.
INTRODUÇÃO
Os catastróficos efeitos da pandemia causada pela doença do SARS-CoV-2 2019 (COVID-19) concentraram os esforços do campo científico no estudo da fisiopatologia referente à infecção pelo vírus, seus efeitos sobre o organismo, seus fatores de risco e a resposta imune envolvida nesse processo1.
Nesse contexto, o reconhecimento dos mecanismos de entrada do vírus, como os receptores Toll-Like 4 (TLR-4) e enzima conversora de angiotensina 2 (ECA 2) tem grande importância para a compreensão da síndrome respiratória aguda grave (SRAG). Ademais, a interação desses receptores com os fatores de risco tem grande impacto na mortalidade de pacientes que venham a desenvolver a doença2,3.
Desde dezembro de 2019, com o início da descrição dos primeiros casos, a presença de doenças crônicas vasculares e/ou metabólicas, como obesidade, diabetes, aterosclerose, hipertensão, além de idade avançada e sexo masculino, tem sido descrita como fator de risco clássico3.
Mais recentemente, com o surgimento de novas variáveis, uma faixa etária mais jovem associada à obesidade tem se destacado4. Além de novas variantes, uma provável suscetibilidade genética tem se revelado como fatores de pior prognóstico nas formas graves da COVID-193,4.
O surgimento constante de novas evidências leva à construção de diferentes hipóteses e levantam mais questionamentos, dentre eles: (a) quais mecanismos o SARS-CoV-2 utiliza para infectar o organismo; (b) qual a relação entre os receptores TLR-4 e ECA 2 e as manifestações típicas da doença; (c) por que os fatores de risco favorecem um prognóstico mais reservado2,3.
Certamente, a imunometabiologia tem um papel relevante na resposta inflamatória do hospedeiro, e esse artigo se dispõe a discutir os principais mecanismos envolvidos nesse processo.
O PAPEL DOS RECEPTORES TOLL-LIKE E DA IMUNIDADE INATA NA COVID-19
Com a finalidade de diferenciar o que é próprio do que não é próprio e manter a homeostasia, o sistema imunológico age através de mecanismos inatos e adaptativos3. Dentre os mecanismos inatos, estão as barreiras físicas, químicas e biológicas, além de células fagocíticas (macrófagos, neutrófilos, células dendríticas e células natural killer - NK), mastócitos e proteínas sanguíneas (sistema complemento e citocinas)3.
Para efeito didático, pode-se dizer que o sistema imune inato constitui a primeira linha de avaliação do organismo do que pode ou não ser tolerado. Isso se dá por meio do reconhecimento de determinados padrões moleculares e construindo uma resposta imunológica rápida através da produção e liberação de interferons, citocinas e quimiocinas pró-inflamatórias quando algum agente é identificado como estranho à homeostase3,5.
Nesse contexto, os receptores Toll-Like (TLR) - da imunidade inata - constituem uma classe de receptores de reconhecimento de padrões (RRPs). Eles se mostram particularmente importantes na ligação e reconhecimento dos padrões moleculares associados a patógenos (PAMPs) e na subsequente ativação da resposta inflamatória. Estão universalmente presentes na membrana ou interior das células imunitárias, principalmente macrófagos, neutrófilos e células dendríticas5.
É importante frisar que os TLR não reconhecem antígenos específicos, mas sim um padrão de moléculas proteicas comumente presentes na superfície de micro-organismos, os PAMPs, e cuja finalidade é tentar deter uma infecção potencial inicial5. Atualmente, já foram identificados mais de 10 tipos de TLR, dentre os quais se destaca o receptor TLR-4. Esse receptor atua no início e na progressão de diversas doenças inflamatórias e infecciosas, além de se ligar fortemente com a glicoproteína Spike do SARS-CoV-2, quando comparado a outros TLR2,6.
A interação entre um TLR e um PAMP desencadeia uma cascata de mecanismos intracelulares. O resultado dessa ativação pode ser a expressão de genes inflamatórios para combater os agentes infecciosos5. Embora cada TLR tenha sua via de sinalização intrínseca e induza uma resposta biológica específica contra determinados patógenos, é importante entender os artifícios comuns relacionados à ativação dos TLR6.
De maneira geral, os TLR possuem domínios extra e intracelulares. Os domínios extracelulares são regiões ricas em leucina que interagem com os PAMPs, enquanto os domínios intracelulares ou citoplasmáticos são homólogos aos domínios de sinalização dos receptores de interleucina-1 (IL-1R) recebendo a designação de domínio Toll/Interleucina-1 (TIR)7.
A ativação dos TLR pela associação a um PAMP age no nível do domínio TIR, recrutando proteínas ativadoras como a MyD88. Essa proteína, por sua vez, age liberando Fator Nuclear kB (NF-kB), o qual é translocado para o núcleo, onde regula-se a expressão de genes relacionados à resposta imune e inflamatória. Dentre esses, temos genes para citocinas, interferons e interleucinas, genes para contrabalancear os processos de apoptose e morte celular e genes que regulam a proliferação e sobrevida celular7.
Os TLR presentes nas membranas plasmáticas (TLR-1, TLR-2, TLR-4, TLR-5 e TLR-6) possuem domínio extracelular e intracelular, enquanto os receptores encontrados nos endossomos (TLR-3, TLR-7, TLR-8, TLR-9 e TLR-10) possuem domínio intracelular6.
Receptor Toll-Like 4 e recepção do SARS-CoV-2 no organismo
O TLR-4 é um dos RRPs mais estudados na atualidade. Além de reconhecer inúmeros PAMPs, identificam-se padrões moleculares associados a danos (DAMPs) resultantes da apoptose, ou morte celular e da injúria tecidual causada, por exemplo, por uma infecção viral2,8.
Outra característica interessante do TLR-4 é que a sua presença não se limita aos macrófagos e aos monócitos do sistema imune, estendendo-se às células de outros tecidos. No pulmão, por exemplo, o TLR-4 pode estar presente nos pneumócitos tipo I e II, nas células do epitélio brônquico e nos macrófagos respiratórios. Já no coração, o TLR-4 se expressa nos cardiomiócitos, nos fibroblastos e nos macrófagos cardíacos2.
Como já explicado anteriormente, a ativação do TLR-4 pelo reconhecimento de determinados padrões moleculares gera uma resposta inflamatória exacerbada através da liberação de mediadores químicos2. Existem duas vias de sinalização envolvendo o TLR-4. Uma delas é a via regular, que culmina na liberação do NF-kB e na subsequente transcrição de genes para expressão de citocinas e quimiocinas pró-inflamatórias, especialmente Fator de Necrose Tumoral-α (TNF-α), Interleucina 1 (IL-1β) e Interleucina 6 (IL-6). A outra é a via alternativa, que utiliza os adaptadores TRIF (do inglês TIR-domain containing adapter-inducing IFN-β) e TRAM (TRIF-related adaptor molecule) e resulta na liberação de Interferon tipo 1 (IFN-1)2.
A via regular também pode ser chamada de via MyD88 dependente e apresenta uma resposta inflamatória mais intensa, formando a "tempestade de citocinas". Já a via alternativa seria MyD88 independente, cursando com uma resposta inflamatória moderada2.
Estudos demonstraram que o receptor TLR-4 é o TLR que forma a ligação proteína-proteína mais forte com a glicoproteína Spike (proteína S) do SARS-CoV-2. Isso pode estar relacionado aos mecanismos de infecção do organismo pelo SARS-CoV-2 e à inflamação generalizada que ocorre nos estágios mais avançados da COVID-192,8.
É importante lembrar, no entanto, que a proteína S do SARS-CoV-2 liga-se fortemente também à ECA 2. Tal informação vem sendo bem explorada no estudo da fisiopatologia do COVID-19, inclusive na relação entre TLR-4 e ECA 22,8.
Acoplamento do vírus e relação do receptor Toll-Like 4 com ECA 2
O SARS-CoV-2 liga-se aos receptores humanos de ECA 2 através da proteína viral Spike, inserindo seu material genético dentro das células hospedeiras e iniciando o processo de replicação viral3. Ao entrar no corpo, o coronavírus infecta os tecidos ricos em ECA 2 (pulmão, epitélio do intestino delgado e endotélio vascular), precipitando os sintomas virais comuns (febre, tosse seca, fadiga e até sintomas gastrointestinais) decorrentes da dominância da patogenicidade viral3.
Neste momento, a doença pode ter um curso benigno e autolimitado, pela contenção por macrófagos, monócitos, citocinas e linfócitos B e T primários, ou evoluir para a fase pulmonar e inflamatória, quando há uma resposta imunológica e inflamatória disfuncional e exacerbada. Essa última pode guardar uma relação com os TLRs, especialmente o TLR-43,8.
Aboudounya e Heads mostraram em um artigo de revisão que, em pacientes com COVID-19, a expressão de TLR-4 e de seus mediadores químicos encontrava-se significativamente aumentada em células sanguíneas mononucleares quando comparada a indivíduos saudáveis. Além disso, a maior liberação de DAMPs pela lise e morte celular causada pela infecção viral é descrita como um outro possível mecanismo de ativação dos TLR-4, principalmente nos pulmões e no coração. Isso acaba por influenciar a ocorrência de processos de inflamação e fibrose2.
A ativação dos TLR-4, no entanto, não é totalmente maléfica. Ela se mostra importante na liberação, principalmente, de interferon tipo β (IFN-β), que age reduzindo a inflamação cardíaca e a replicação viral. Todavia, a hiperestimulação deste receptor pode levar a um estado inflamatório grave, como citado anteriormente2.
Ainda em sua revisão, Aboudounya e Heads propuseram um modelo em que o SARS-CoV-2 infecta primeiramente o epitélio pulmonar, através da ligação entre a proteína S viral e o receptor ECA 2 presentes nos pneumócitos tipo II. Esse processo leva à apoptose e morte celular, à diminuição da produção de surfactante nos alvéolos pulmonares e ao comprometimento do sistema respiratório, podendo causar, em estágios mais avançados, a SRAG2.
Logo em seguida, há a ativação do TLR-4 por dois mecanismos: (a) o acoplamento do vírus ao domínio extracelular do TLR-4; (b) a liberação de DAMPs provenientes da injúria tecidual. O SARS-CoV-2, ao se acoplar ao TLR-4, pode gerar um aumento da expressão de ECA 2, infectar diretamente a célula, ou aumentar a ativação da via de sinalização MyD88 dependente, pró-inflamatória, em detrimento da MyD88 independente, a qual libera IFN-β e possui função antiviral e cardioprotetora2.
A estimulação do TLR-4 pelos DAMPs está também relacionada ao aumento do risco de trombose, infarto do miocárdio e embolia e, com o tempo, essa estimulação pode causar fibrose no coração e pulmões2.
Podemos concluir, portanto, que a elucidação do papel desses dois importantes receptores no desenvolvimento da patologia causada pelo novo coronavírus é essencial para o avanço na triagem, tratamento e melhora do prognóstico dos pacientes que venham a contrair a doença.
IMPORTÂNCIA DOS RECEPTORES ECA 2 NO ORGANISMO
A ECA 2 é uma metaloprotease composta por 805 aminoácidos que possui certa semelhança com a enzima conversora de angiotensina 1 (ECA 1), porém com funções diferentes9. Ela atua como uma carboxipeptidase e está abundantemente disseminada pelo organismo, notadamente em células epiteliais alveolares, cardíacas, endoteliais, renais e pancreáticas9.
A ECA 2 tem como função contrarregular o sistema renina-angiotensina-aldosterona (SRAA), por agir de forma antagônica à ECA 1 (Figura 1)10,11. Além disso, possui função protetora contra danos respiratórios, cardíacos, renais, endoteliais e hepáticos9.
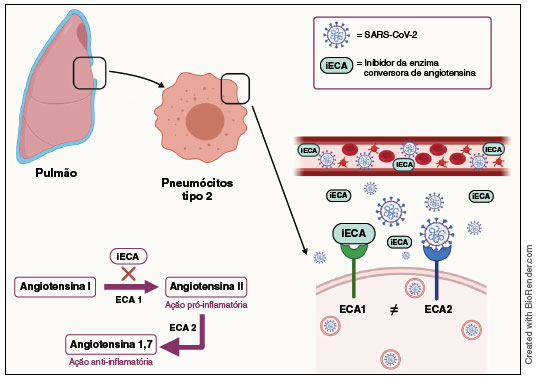
Figura 1
Ações opostas das enzimas conversoras da Angiotensina (ECA) 1 e 2 (ECA 2), presentes em vários órgãos do corpo, porém, ilustradas nesta imagem nas células pulmonares. O efeito patogênico do novo coronavírus (SARS-CoV-2) é contrário ao das drogas iECA. Através da sua ligação à ECA 2, ele reduz as ações desta enzima e, assim, ativa indiretamente o sistema renina-angiotensina-aldosterona. A droga iECA, por sua vez, bloqueia a ação da ECA 1 e reduz a formação de angiotensina II
Sua ação se dá pela transformação da Angiotensina 2 (Ang II), forma ativa da Angiotensina 1 (Ang I), em Angiotensina 1-7 (Ang 1-7)9,10. A Ang II é um importante vasoconstritor que age principalmente nas arteríolas. Com o objetivo de aumentar a pressão arterial, ela ainda atua diretamente no rim, estimulando a troca de sódio por hidrogênio no túbulo proximal renal, e aumenta a reabsorção de sódio e de bicarbonato12,13. Adicionalmente, a Ang II age no hipotálamo aumentando a secreção do hormônio antidiurético para estimular a reabsorção de água nos ductos coletores renais e induzir maior sensação de sede, para maior ingestão de água14.
Para a Ang II ser capaz de realizar suas funções biológicas, ela precisa, inicialmente, se ligar a seus receptores AT1 ou AT2. Geralmente, a Ang II se liga ao receptor AT1 formando o complexo Ang II/AT113. Esse complexo gerará efeitos como vasoconstrição, inflamação, fibrose tecidual e disfunção do endotélio, ou seja, ações mais pró-oxidativas e pró-inflamatórias que, por sua vez, podem ser maléficas e gerar danos significativos ao organismo se superativadas11,15. Já o complexo Ang II/AT2 age como vasodilatador, por meio da liberação de óxido nítrico e bradicinina, e é mais encontrado em tecidos embrionários, tendo expressão limitada à glândula adrenal, endotélio vascular, cérebro, rim e ovário16.
Dessa forma, quando a ECA 2 degrada a Ang II (ativa) em Ang 1-7 (inativa) consegue diminuir a ação inflamatória e oxidativa, por meio da formação do complexo Ang 1-7/Receptor Mas (MasR). Esse complexo age de forma a suprimir mecanismos que geram exacerbação da inflamação e fibrose tecidual, por meio da redução da migração de leucócitos, regulação negativa de citocinas pró-inflamatórias e redução dos fatores de fibrose tecidual16.
O complexo Ang 1-7/MasR estimula a vasodilatação, por meio da liberação de bradicinina e grelina, e atenua fatores geradores de vasoconstrição. Ele gera a neutralização de processos inflamatórios da Ang II através da inibição da enzima óxido nítrico sintase induzível (iNOS), oxidase não fagocítica NADPH (Nox) e vias de sinalização pró-inflamatórias17. Além do mais, a ECA 2 também diminui a ação de algumas citocinas, como o TNF-α e a IL-6, que promovem a inflamação sistêmica ou local e atraem células fagocitárias, principalmente os macrófagos, para os sítios de ativação10.
O novo coronavírus e sua relação com os receptores ECA 2
O coronavírus é um vírus da ordem Nidoviralez e da família Coronaviridae, que, normalmente, infecta animais e, por isso, é classificado como zoonótico. Esta é uma família que causa infecções respiratórias e recebe o nome coronavírus por possuir projeções no seu envelope, em forma de espículas, formadas pela proteína S, lembrando um aspecto de coroa18.
Até hoje, sete tipos de coronavírus foram descobertos: alfa coronavírus HCoV-229E, alfa coronavírus HCoV-NL63, beta coronavírus HCoV-OC43, beta coronavírus HCoV-HKU1, SARS-CoV, do inglês, severe acute respiratory syndrome - coronavirus, MERS-CoV, do inglês, Middle East Respiratory Syndrome - coronavírus, e o SARS-CoV-2 (novo coronavírus), do inglês Severe Acute Respiratory Syndrome - coronavírus-2 18,19.
Os coronavírus 229E, NL63, OC43 e HKU1 estão associados a doenças com sintomatologia leve. Porém, os coronavírus SARS-CoV, MERS-CoV e SARS-CoV-2 podem gerar doenças respiratórias graves nos humanos, muitas vezes fatais19.
O SARS-CoV-2 é um betacoronavírus com material genético composto por uma fita simples de ácido ribonucleico (RNA). Ele possui até 30.000 nucleotídeos em todo o seu genoma que são formados por uma base nitrogenada (guanina, adenina, citosina, uracila), uma ribose (molécula de açúcar) e um ácido fosfórico19. Esse vírus, por ser RNA+, pode ser lido facilmente pelos componentes celulares. Assim, nas células humanas infectadas, ele age como um RNA mensageiro, conseguindo facilmente se expressar geneticamente19. Ao entrar em contato com ribossomos intracelularmente, o SARS-CoV-2 induz a produção de proteínas virais. De início, são produzidas algumas enzimas, dentre elas um complexo replicase-transcriptase e uma RNA polimerase, que serão essenciais para a construção de um RNA-, usando como base o RNA+ do vírus19.
Esse RNA- serve como modelo para a produção de outras moléculas de RNA+, idênticas ao RNA+ original. Todo esse processo ocorre nos endossomos, compartimentos membranosos encontrados no citoplasma celular. No final, estão formados novos vírus e seus componentes proteicos19. Esses componentes são os fatores que estruturam os vírus: proteína E do envelope, proteína M da membrana, proteína N do nucleocapsídeo e proteína S das espículas que formam a coroa (Figura 2)2.
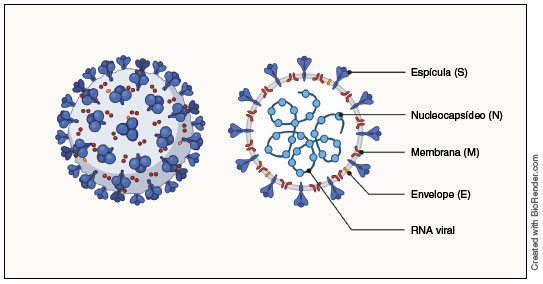
Figura 2
Estrutura do vírus SARS-CoV-2 com seus componentes proteicos
A proteína S está presente como um trímero em vírus maduros e possui dois principais componentes, as proteínas S1 e S2. A proteína S1 do SARS-CoV-2 se liga perfeitamente aos receptores de membrana ECA 2 (Figura 1)16 após sua ativação pela enzima proteolítica serina 2 transmembrana protease (TMPRSS2). Desta forma, os vírus conseguem adentrar no citoplasma com a fusão entre as membranas viral e celular16,19.
A doença do SARS-CoV-2 se caracteriza por uma menor disponibilidade desses receptores ECA 2 (fenômeno de hiporregulação), uma vez que eles estão sendo ocupados pelos vírus. Isto gera uma maior suscetibilidade do organismo a danos sistêmicos, pela maior formação do complexo Ang II / AT1 devido à diminuição da ECA 2/Ang 1-7 (Figura 1)10. A falta da ECA 2 pode também aumentar o número de macrófagos e potencializar a ação de citocinas pró-inflamatórias, o que agrava ainda mais o estado hiperinflamatório da COVID-1910. Esta desregulação inflamatória pode ser ainda maior em indivíduos com doenças cardiometabólicas de base.
INFLAMAÇÃO SUBCLÍNICA DA OBESIDADE E COVID-19
A obesidade é uma doença crônica não transmissível (DCNT), caracterizada pelo acúmulo excessivo de gordura corporal, que aumenta o risco para o surgimento de diabetes, hipertensão, aterosclerose e câncer. Vale ressaltar que as possíveis complicações relacionadas à obesidade vão variar de acordo com o grau de excesso de gordura e sua localização corpórea20, sendo importante considerar onde a gordura corporal está depositada, se na tela subcutânea ou intra-abdominal (visceral). Essa distinção é de suma importância, uma vez que há diferenças entre ambas em relação à sua celularidade, atividade, função endócrina e resposta à insulina21.
O tecido adiposo visceral é metabolicamente mais ativo que o tecido adiposo subcutâneo21. Logo, produz mais adipocinas inflamatórias, como a leptina, e menos adiponectina, que possui propriedades anti-inflamatórias e de melhora na resistência à insulina. As adipocinas são proteínas secretadas pelo tecido adiposo e apresentam funções metabólicas, como adipogênese, homeostase de energia, controle do apetite, imunidade e inflamação, assim como homeostase glicêmica e sensibilidade à insulina, culminando na inflamação subclínica da obesidade22.
A leptina tem um papel importante no imunometabolismo em pacientes obesos, desde que seus receptores foram descritos nas células da imunidade inata e adquirida. Pacientes com obesidade de predomínio visceral têm maior número de macrófagos com fenótipo inflamatório, além de mobilizarem mais ácidos graxos livres (AGL) pela lipólise23,24.
Segundo a Organização Mundial de Saúde, a prevalência mundial da obesidade aumentou cerca de três vezes desde 1975, sendo os adultos a faixa etária mais acometida25,26. Atualmente, essa comorbidade também vem sendo classificada como um preditor importante para complicações graves da COVID-19. Uma pesquisa realizada por Peng (2020) evidenciou que pacientes com índice de massa corporal (IMC) acima do ideal, ou seja, igual ou superior a 30 kg/m2, foram associados a uma maior frequência de casos críticos de COVID-19 quando comparados aos pacientes com IMC normal27-29.
Nos Estados Unidos, país onde 40% da população é obesa, estudos avaliaram que cerca de mais de 50% dos adultos confirmados com COVID-19 apresentavam obesidade e, destes, cerca de 11,7% foram a óbito, o que mostrou a relação entre maior IMC e mortalidade, particularmente em pacientes até os 65 anos30.
A COVID-19 não é a primeira infecção viral respiratória agravada pelo excesso de peso. A pandemia do H1N1 de 2009 também foi intensificada por este fator25. Os pacientes obesos apresentam-se com fator de risco à frente de outras doenças, tendo em vista a diminuição na imunidade adaptativa devido à alteração na função das células T e B e também na imunidade inata. O desequilíbrio na produção das adipocinas pró e anti-inflamatórias aumenta a proliferação e responsividade das células Th-1, Th-2, Th-17, e reduz a resposta do Treg. Na imunidade, aumenta o fenótipo de macrófagos M1, células NK e dendríticas. Assim, liberam uma quantidade elevada de citocinas inflamatórias, sendo as principais IFN-gama, TNFα, IL-1 e a IL-625.
Dessa forma, em pacientes obesos, os níveis elevados de TNF-α evidenciam a importância do seu papel na citotoxicidade da resposta imunológica, além de contribuir na resistência à insulina (RI) e diabetes, por inibir a fosforilação da tirosina presente no substrato-1 do receptor de insulina (IRS-1). Além disso, por um lado a IL-1 contribui para a formação do fator de transcrição NF-kB, estimulando a superexpressão da sinalização inflamatória do fator de crescimento endotelial vascular (VEGF). Por outro, a IL-6 atua favorecendo os distintos tipos de câncer associados à inflamação21,25.
As principais consequências causadas por esse processo inflamatório crônico são a hipóxia e isquemia. Por sua vez, ambas levam a um estado de estresse oxidativo, estimulando ainda mais a secreção dessas proteínas inflamatórias e radicais reativos de oxigênio, que danificam a funcionalidade mitocondrial e do DNA25.
O aumento primário da resposta inflamatória específica da obesidade é exacerbado após a infecção pelo SARS-CoV-2, quando células mieloides, como os macrófagos e monócitos, são recrutadas por quimiocinas fabricadas por células epiteliais infectadas. Os monócitos comprometidos vão produzir níveis elevados de IFNs, responsáveis por induzir a ECA 2 e citocinas pró-inflamatórias, sendo fator desencadeante para o estado hiperinflamatório evidenciado na COVID-19 grave, caracterizada pela "tempestade de citocinas"31. Tal condição leva a um pior prognóstico, propiciando o quadro de insuficiência respiratória e falência múltipla de órgãos10.
O elo molecular entre a ECA 2 e o SARS-CoV-2 pode representar também um fator essencial na relação entre a gravidade de COVID-19 e RI, caracterizada pela diminuição na capacidade da insulina de realizar a sua função de manter a manutenção da homeostase de glicose. Por conseguinte, esse mecanismo contribui para elevar os níveis glicêmicos, os quais estão associados à capacidade de replicação viral, uma vez que a hiperglicemia está relacionada a menor capacidade de resposta imune31.
A RI apresenta vínculo direto com a adiposidade, principalmente nos pacientes com obesidade visceral, devido ao aumento da atividade nervosa simpática, responsável por promover uma ativação do SRAA26. A ECA 2 está presente em maior quantidade no tecido adiposo, que é visto como um reservatório viral26. Nos adipócitos, ela limita a expressão de citocinas pró-inflamatórias, atuando como uma via protetora com muita relevância contra doenças pulmonares, diabetes mellitus e hipertensão. Porém, nos casos de COVID-19, a ECA 2 passa a atuar como porta de entrada para o SARS-CoV-2. Neste cenário, teremos uma menor disponibilidade destes receptores celulares e assim uma hiporregulação desta enzima32.
A atenuação funcional da ECA 2 compromete a formação de Ang 1-7, responsável por melhorar o metabolismo da glicose e a RI, uma vez que ela atua minimizando os efeitos inibitórios da Ang II na captação celular de glicose. Além disso, a perda de ECA 2 impede a neutralização do eixo Ang II/AT1, o qual é ativado em condições patológicas. Logo, com a presença do SARS-CoV-2, esse eixo estará atuante, aumentando a sua atividade na secreção e sinalização da insulina e absorção da glicose, bem como na inflamação que conduz ao desenvolvimento da RI33, (Figura 3).
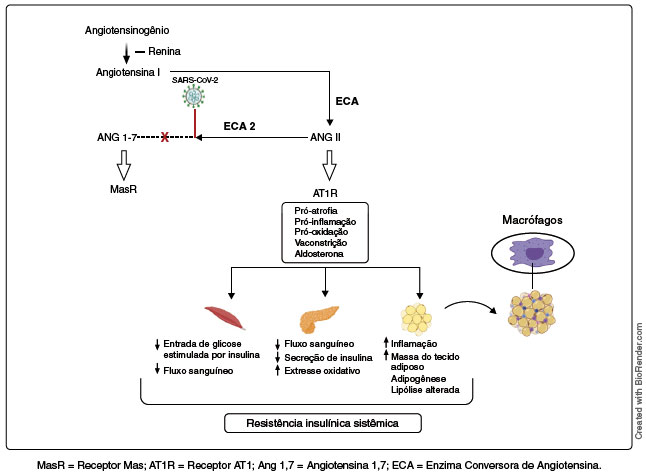
Figura 3
Ações da angiotensina II. Na COVID-19, há uma redução e inibição da ECA 2, pois ela serve como reservatório do SARS-CoV-2, levando a reações pró-inflamatórias sistêmicas no músculo esquelético, pâncreas e tecido adiposo. Os desequilíbrios provocados pela ação da angiotensina II (Ang II) contribuem para a resistência insulínica sistêmica
A ECA 2 é expressa em vários tecidos, incluindo músculo esquelético, células beta-pancreáticas e tecido adiposo. Logo, sua alteração no SRAA irá provocar repercussões em todos esses locais, contribuindo para a RI. No músculo esquelético, os impactos da Ang II na captação da glicose estão pautados nas alterações no fluxo sanguíneo local, assim como déficit na sinalização de insulina e absorção de glicose estimulada pela redução da GLUT-4 (GLUcose Transporter type-4), levando ao aumento de glicose plasmática e bloqueio de receptor de insulina. No pâncreas, o Ang II também atua diminuindo o fluxo sanguíneo pancreático e a secreção de insulina produzida pelas células beta-pancreáticas. Além disso, esse aumento na atividade de SRAA contribui para a inflamação e fibrose pancreática34.
No tecido adiposo, os receptores de Ang II foram descritos com capacidade de alterar a adipogênese, ou seja, a diferenciação dos pré-adipócitos em adipócitos. Essas células encontram-se hipertróficas e, consequentemente, tornam-se mais lipolíticas, aumentando a concentração de AGL no plasma, levando à lipotoxicidade. Os adipócitos maduros apresentam uma maior proteção contra esse efeito lipotóxico devido à sua alta capacidade de detoxificação de ácidos graxos. Em contrapartida, os pré-adipócitos são mais sensíveis e, especialmente neles, os AGL circulantes ativam proteínas TLR-4 estimulando a ativação de vias inflamatórias que também irão interferir na captação de glicose pela sinalização da insulina22.
Aminian Ali e Tu Chao, em sua metanálise, observaram a associação entre cirurgia bariátrica e metabólica como um fator responsável por reverter os impactos mecânicos e fisiológicos negativos da obesidade, assim como a repercussão da RI, funcionando como agente protetor contra a forma grave da infecção pelo SARS-CoV-2. Por isso, pacientes obesos que realizaram esse procedimento apresentaram três vezes menos chances de ir a óbito, comparado com os pacientes sem cirurgia bariátrica anterior, evidenciando um melhor desfecho na associação entre COVID-19 e obesidade35.
DOENÇA ATEROSCLERÓTICA, COVID-19 E ECA 2
No quadro de COVID-19 existe um maior recrutamento de células imunológicas que, por agressão direta ao endotélio ou por resposta imunomediada, pode ocasionar o agravamento da disfunção endotelial causada pela aterosclerose. Esta promove uma instabilidade da placa e também a formação de trombos3.
O desequilíbrio do SRAA é ocasionado pela ligação do SARS-CoV-2 aos receptores da ECA 2. Como a enzima promove a degradação da Angiotensina II em Angiotensina 1-7, quando o vírus se liga, contribui para o aumento da pressão arterial e do estado pró-inflamatório, já presente na doença aterosclerótica, o que causa uma maior disfunção endotelial no paciente36.
Alguns estudos mostram que a Ang II induz a produção de mediadores importantes para a gênese da disfunção endotelial e, consequentemente, a formação das placas ateroscleróticas, como MCP-1 (Proteína 1 quimioatraente de monócitos), VCAM-1 (Proteína 1 de adesão celular vascular), IL-6, TNF-α, MMP-2 (Metaloproteinase matriz do tipo 2) e MMP-9 (Metaloproteinase matriz do tipo 9)37,38. Por isso, o desequilíbrio ocasionado pelo SARS-CoV-2 na biodisponibilidade dos receptores ECA 2 ocasiona uma piora do quadro inflamatório da aterosclerose.
A resposta anti-inflamatória mediada pelo ECA 2 no quadro de aterosclerose está relacionada com a produção de Ang 1-739. Além disso, o receptor ECA 2 da célula endotelial está relacionado com a diminuição do estresse oxidativo, que apresenta ligação com a patogênese da aterosclerose40.
Alguns estudos em camundongos submetidos a nocaute gênico demonstram que a deficiência do receptor ECA 2 aumenta a probabilidade de desenvolvimento de aterosclerose41,42. Em contrapartida, a superexpressão gênica da ECA 2 em camundongos levou a uma diminuição do tamanho da lesão aterosclerótica dentro do seio aórtico, em comparação ao grupo controle43.
A disfunção endotelial é uma parte essencial da patogênese da COVID-19 grave, ocasionando a disfunção de múltiplos órgãos, insuficiência respiratória e trombose44. Por isso, é de se esperar que, em pacientes mais idosos, a lesão endotelial causada pela infecção do SARS-CoV- 2 seja exacerbada devido a uma inflamação pré-existente nesses pacientes, pela maior população senescente de adipócitos e pelas mudanças das células T associadas à idade, as quais são responsáveis pela amplificação das respostas inflamatórias subsequentes45.
HIPERTENSÃO E COVID-19
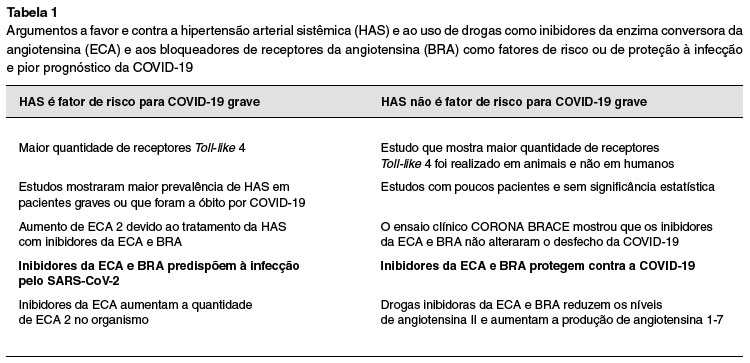
A hipertensão arterial sistêmica (HAS) tem sido associada a um maior risco de COVID-19 grave46-48. Em uma metanálise com seis estudos, totalizando 1527 pacientes com COVID-19, cerca de 17% tinha hipertensão. Quando se comparou a frequência de HAS de acordo com a gravidade da doença, ela foi cerca de duas a três vezes mais frequente nos pacientes em estado grave do que naqueles com COVID-19 leve49.
Um outro estudo mostrou uma prevalência de HAS de 58% em pacientes com infecção grave por COVID-1947. Essa alta prevalência de HAS em pacientes com COVID-19 grave levanta a questão sobre uma maior susceptibilidade para infecção ou para desfechos mais graves nesses indivíduos hipertensos49-54.
Desde o início da pandemia do SARS-CoV-2, várias publicações e estudos têm relatado maior risco de COVID-19 grave naqueles com doenças cardiovasculares e metabólicas8,46,55,47-54. Chen e cols. compararam dados clínicos de 113 pacientes falecidos por COVID-19 com 161 recuperados da doença. A média de idade dos que faleceram foi de 68 anos versus 51 anos dos sobreviventes. Neste estudo, 73% dos que foram à óbito eram do sexo masculino versus 55% no grupo de sobreviventes. Outro dado que chamou a atenção foi a maior prevalência de HAS e outras doenças cardiovasculares nos falecidos, 48% versus 24%, respectivamente55.
Na fisiopatologia da HAS, diversos mecanismos levam à desregulação do SRAA, como aumento da pressão dos capilares glomerulares, disfunção endotelial, elevação dos níveis de citocinas pró-inflamatórias e radicais livres. A disfunção endotelial se dá pela redução da biodisponibilidade do óxido nítrico (NO), resultado da elevação de Ang II, o que acarreta vasoconstricção e dano vascular8,56. Além disso, estudos em animais hipertensos mostraram uma maior expressão de receptores TLR-457. Todas essas alterações fisiopatológicas da HAS podem explicar a maior susceptibilidade de indivíduos hipertensos à forma grave da COVID-19.
Outra metanálise ressaltou também esta hipótese, uma vez que a HAS esteve associada a um risco cerca de 2,5 vezes maior de infecção grave ou morte pela COVID-19, especialmente nos mais idosos50.
Adicionalmente, considerando a desregulação do SRAA presente tanto na HAS quanto na infecção pelo SARS-CoV-2, o tratamento com drogas hipotensoras das classes dos inibidores da enzima conversora de angiotensina (iECA) e os bloqueadores do receptor de angiotensina (BRA) tem sido avaliado no contexto da COVID-19. Enquanto o SARS-CoV-2 reduz a disponibilidade da ECA 2, os iECAs agem na ECA 1, enzimas com funções e sítios de ação diferentes (Figura 1).
Há dados limitados quanto a uma possível ação dos iECAs e BRAs em aumentar a expressão da ECA 2 nas membranas celulares, o que poderia acarretar uma maior susceptibilidade para o adoecimento e para casos mais graves da COVID-1949,52-54,58. No entanto, esses resultados são com altas doses destes medicamentos e variam a depender da classe da droga e do órgão analisados50-52. Outra hipótese aventada é que essas drogas sejam na realidade protetoras, uma vez que elas bloqueiam a ação da Ang II, neurormônio sabidamente desregulado na COVID-19 grave53,58.
Estudo randomizado brasileiro, o BRACE CORONA, objetivou responder este questionamento, se haveria diferenças nos desfechos de pacientes hospitalizados com COVID-19 ao se suspender ou manter o uso de iECA ou BRA durante o internamento. O estudo englobou 659 pacientes, 40% mulheres, idade média 55 anos, em uso crônico de iECA ou BRA. Não foram observadas diferenças entre os grupos em termos de mortalidade e dos demais desfechos analisados. A taxa de morte foi de 2,7% no grupo em que se suspendeu as drogas versus 2,8% naqueles que continuaram recebendo os medicamentos58,59.
Em conclusão, ainda não há dados suficientes para determinar com mais certeza se o descontrole da pressão arterial é realmente um fator de risco tanto para maior susceptibilidade à infecção pelo SARS-CoV-2, quanto para pior prognóstico da COVID-19. Os estudos publicados apresentam limitações quanto à verificação das comorbidades, tendo em vista que a grande maioria consiste em informações coletadas na admissão hospitalar e não por um acompanhamento médico periódico. Também há vários estudos que possuem um número limitado de pacientes analisados, o que dificulta a obtenção de dados estatisticamente significativos (Tabela 1)52.
DESREGULAÇÃO POSITIVA E NEGATIVA DA ECA 2 NA COVID-19
A partir da análise de que pacientes com fatores de risco para SARS-CoV-2, como idade avançada, hipertensão, diabetes, obesidade e doença cardiovascular, apresentaram maior gravidade da COVID-19, a hipótese de que essas condições favoreçam à maior gravidade da COVID-19 devido a uma deficiência basal de ECA 2 foi levantada. Verdecchia e cols. concluíram que a regulação negativa de ECA 2 induzida pelo SARS-CoV-2 pode ser especialmente prejudicial em pessoas com deficiência basal de ECA 2, uma vez que os níveis dessa enzima seriam ainda mais reduzidos na COVID-19, e assim teríamos um maior efeito deletério da Ang II60.
Entretanto, outros pesquisadores aventam que condições com aumento de ECA 2 nos tecidos induziriam uma maior gravidade na COVID-19, como nos casos do fumo e da doença pulmonar obstrutiva crônica (DPOC)61. Em estudo feito por Russo e cols., os autores concluíram que a nicotina seria um fator de risco para COVID-19, uma vez que seu uso prolongado causaria a regulação positiva da ECA 2, principal receptor utilizado para o SARS-CoV-2 se ligar à célula, através da α7-nAChRs62. Apoiando essa hipótese, foi proposto por Leung e cols., então, a administração de antagonistas de α7-nAChR para diminuir a expressão de ECA 2 nesses pacientes e evitar a infecção pelo SARS-CoV-2.
Entretanto, pesquisas foram feitas em relação a essa hipótese de aumento de ECA 2 nessa população. A partir de um extenso estudo comparativo entre a proporção estimada de tabagistas e quantidade de pacientes com COVID-19 hospitalizados, concluiu-se que houve uma baixa prevalência de fumantes entre os hospitalizados em comparação com o esperado se houvesse a regulação positiva de ECA 2 e, então, maior gravidade da doença nesses pacientes63.Pelas suas observações, e pela α7-nAChR estar envolvida na modulação da secreção de citocinas pró-inflamatórias e suprimir a tempestade de citocinas64,65, a administração de seu antagonista pode acarretar maior gravidade nos pacientes com COVID-1963.
Em relação ao SRAA, as suspeitas iniciais de que diferentes componentes do sistema estariam alterados nos pacientes com coronavírus foram descartadas através do estudo de Rojas e cols., em 2020. Ao contrário do que se esperava, a quantidade de ECA 2 solúvel (ECA 2s) nos pacientes infectados não foi alterada em relação aos pacientes saudáveis. Além dos níveis absolutos, a atividade de ECA 2s dos pacientes graves infectados pelo SARS-CoV-2 um mês após recuperação clínica demonstrou-se inalterada66.
A ECA 2 é encontrada originalmente nas células do músculo cardíaco, fibroblastos cardíacos, endotélio vascular coronário, rim, fígado, intestino delgado, testículos, cérebro, células epiteliais alveolares do pulmão, linfócitos na mucosa oral, enterócitos, células endoteliais arteriais e venosas, e células do músculo liso arterial. Apesar disso, pequena quantidade de sua forma solúvel foi detectada no plasma dos pacientes com coronavírus no estudo de Mehrabadi e cols.16.
Essa forma solúvel da enzima foi proposta como um possível biomarcador para análise de gravidade da COVID-19, por estar relacionada à carga viral do paciente66. Essa teoria, porém, não foi corroborada pelos dados obtidos por Rojas e cols. (2020). Assim como a quantidade de ECA 2s não influencia na gravidade da doença, também foi contrário ao uso rotineiro de ECA 2s no acompanhamento de pacientes com coronavírus66.
Outro ponto a respeito da ECA 2s é a hipótese de ser utilizada como uma forma de terapia para a COVID-19. Apesar do aumento da ECA 2 ser ainda um ponto discutido pelos cientistas no que se refere a uma maior gravidade de doença, a administração de ECA 2s como uma proteína ECA 2s recombinante poderia ser uma alternativa. Isso porque a ECA 2s recombinante poderia ligar-se ao SARS-CoV-2 e, assim, bloquear a ligação do SARS-CoV-2 nas outras ECA 2 dos tecidos67. Uma outra alternativa hipotética seria a administração de ECA 2s modificada que, ao ligar-se ao vírus, neutralizaria a proteína S, inibindo sua entrada. Isso porque a ECA 2s poderia apresentar uma proteína de fusão rhECA 2 de ligação de alta afinidade ao domínio de ligação ao receptor de SARS-CoV-268.
CONCLUSÕES
A fisiopatologia da COVID-19 grave está relacionada a diversos fatores, como obesidade, diabetes, hipertensão, aterosclerose, idade avançada, carga viral no momento da infecção e a genética do indivíduo. A infecção pelo SARS-CoV-2 é responsável por uma menor biodisponibilidade de receptores ECA 2 e uma maior ativação de receptores TLR-4, os quais já estão previamente hiperestimulados em situações específicas. Isso culmina por agravar diversas patologias pré-existentes, como discutido anteriormente.
Dessa forma, o controle de condições pré-existentes à infecção se torna um dos principais alvos terapêuticos para a prevenção do agravamento da COVID-19. Contudo, diante da dificuldade do controle da pandemia e das lacunas no conhecimento do novo coronavírus e seu comportamento biológico, faz-se necessária a elaboração de novos estudos. Haja vista a recente observação de que a idade avançada, outrora fator de risco relevante, e a observação de casos graves em indivíduos adultos jovens, sobretudo associada à obesidade, é necessário o entendimento da interação entre o sistema imune e metabólico. O receptor ECA 2 responde muitos desses questionamentos, mas ainda não se sabe qual a principal via responsável por deflagrar a tempestade de citocinas. O conhecimento permitirá a elaboração de medicamentos que atuem em alvos imunológicos terapêuticos e preventivos da COVID-19, além das vacinas.
REFERÊNCIAS
1. Seyed Hosseini E, Riahi Kashani N, Nikzad H, Azadbakht J, Hassani Bafrani H, Haddad Kashani H. The novel coronavirus Disease-2019 (COVID-19): Mechanism of action, detection and recent therapeutic strategies. Virology. 2020;551:1-9.
2. Aboudounya MM, Heads RJ. COVID-19 and Toll-Like Receptor 4 (TLR4): SARS-CoV-2 May Bind and Activate TLR4 to Increase ACE2 Expression, Facilitating Entry and Causing Hyperinflammation. Mediators Inflamm. 2021;2021.
3. Brandão SCS, Silva EAGBB, Ramos JOX, Melo LMMP, Sarinho ESC. COVID-19, imunidade, endotélio e coagulação: compreenda a interação. Recife; 2020. p. 70.
4. SARS-CoV-2 Variants Classifications and Definitions. Centers for Disease Control and Prevention, 2021 [Internet]. Disponível em: <https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/scientific-brief-emerging-variants.html>.
5. Cruvinel WM, Júnior DM, Araújo JAP, Catelan TTT, Souza AWS, Silva NP, et al. Sistema Imunitário - Parte I. Fundamentos da imunidade inata com ênfase nos mecanismos moleculares e celulares da resposta inflamatória. Rev Bras Reumatol. 2010;50(4):434-61.
6. Ferraz EG, Silveira BBB, Sarmento VA, Santos JN. Receptores Toll-Like: ativação e regulação da resposta imune. RGO - Revista Gaúcha Odontol. 2011;59(3):483-90.
7. Bártholo RM, Bártholo TP. Imunidade inata e a importância dos receptores Toll-similar. Pulmão RJ [Internet]. 2009;Supl 2:S2-S8. Disponível em: http://www.sopterj.com.br/wp-content/themes/_sopterj_redesign_2017/_revista/2009/suplemento-pneumonia/imunidade-inata-e-a-importancia-dos-receptores-toll-similar.pdf
8. Brandão SCS, Ramos JOX, Dompieri LT, Godoi ETAM, Figueiredo JL, Sarinho ESC, et al. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2021 Apr;58:102-10. doi: 10.1016/j.cytogfr.2020.09.002.
9. Francischetti EA, Francischetti A, Abreu VG. A emergência de um novo modulador cardiovascular - A 2a enzima de conversão da Angiotensina 2 (ECA2). Rev SOCERJ. 2005;18(1):36-40.
10. Brandão S, Godoi E, Cordeiro L, Bezerra C, Ramos J, Arruda G, et al. Obesidade e Risco de COVID-19 Grave [Internet]. Repositório Digital da UFPE. 2020. 112 p. Disponível em: https://repositorio.ufpe.br/handle/123456789/37572.
11. Silva TF, Vaz TM, Souza MS, Ferreira, LP, Limborço Filho M, Fontes MAP, et al. O envolvimento do sistema Renina-Angiotensina nas disfunções cardiovasculares e seus recursos farmacológicos. Rev Cient Multidisciplinar Núcleo do Conhecimento. 2019;2(11):181-96. doi 10.32749/nucleodoconhecimento.com.br/saude/renina-angiotensina.
12. Cunha RS, Ferreira AVL. Sistema renina-angiotensina-aldosterona e lesão vascular hipertensiva. Rev Bras Hipertens. 2000;3(3):282-92.
13. Gonsalez SR, Ferrão FM, Souza AM, Lowe J, Morcillo LSL. Inappropriate activity of local renin-angiotensin-aldosterone system during high salt intake: impact on the cardio-renal axis. J Bras Nefrol. 2018;40(2):170-8.
14. Naves LA, Vilar L, Costa ACF, Domingues L, Casulari LA. Distúrbios na secreção e ação do hormônio antidiurético. Arq Bras Endocrinol Metabol. 2003;47(4):467-81.
15. Scholz JR, Lopes MACQ, Saraiva JFK, Colombo FC. COVID-19, renin-angiotensin system, angiotensin-converting enzyme 2, and nicotine: What is the interrelation? Arq Bras Cardiol. 2020;115(4):708-11.
16. Mehrabadi ME, Hemmati R, Tashakor A, Homaei A, Yousefzadeh M, Hemati K, et al. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed Pharmacother. 2021 May;137:111363. doi: 10.1016/j.biopha.2021.111363.
17. Kuriakose J, Montezano AC, Touyz RM. ACE2/Ang-(1-7)/Mas1 axis and the vascular system: vasoprotection to COVID-19-associated vascular disease. Clin Sci. 2021;135(2):387-407.
18. Lima CMAO. Information about the new coronavirus disease (COVID-19). Radiol Bras. 2020;53(2):v-vi. doi: 10.1590/0100-3984.2020.53.2e1.
19. Uzunian A. Coronavirus SARS-CoV-2 and Covid-19. J Bras Patol e Med Lab. 2020;56:1-4.
20. Pinheiro ARDO, De Freitas SFT, Corso ACT. Uma abordagem epidemiológica da obesidade. Rev Nutr. 2004;17(4):523-33.
21. Sippel C, Bastian RMA, Giovanella J, Faccin C, Contini V, Dal Bosco SM. Processos inflamatórios da obesidade. Rev Atenção à Saúde. 2014;12(42):48-56.
22. Jensen MD. Visceral fat: culprit or canary? Endocrinol Metab Clin North Am. 2020;49(2):229-37.
23. Prado WL, Lofrano MC, Oyama LM, Dâmaso AR. Obesidade e adipocinas inflamatórias: implicações práticas para a prescrição de exercício. Rev Bras Med Esporte. 2009;15(5). doi: 10.1590/S1517-86922009000600012.
24. Francisco V, Pino J, Campos-Cabaleiro V, Ruiz-Fernández C, Mera A, Gonzalez-Gay MA, et al. Obesity, fat mass and immune system: Role for leptin. Front Physiol. 2018;9:640. doi: 10.3389/fphys.2018.00640.
25. Michalakis K, Ilias I. SARS-CoV-2 infection and obesity: Common inflammatory and metabolic aspects. Diabetes Metab Syndr Clin Res Rev [Internet]. 2020;14(4):469-71. Disponível em: https://doi.org/10.1016/j.dsx.2020.04.033
26. Petrakis D, Margină D, Tsarouhas K, Tekos F, Stan M, Nikitovic D, et al. Obesity - a risk factor for increased COVID-19 prevalence, severity and lethality (Review). Mol Med Rep. 2020;22(1):9-19.
27. Costa TRM, Correia RS, Silva PHS, Lópes Barbosa GS, Oliveira LM, Cruz VT, et al. A obesidade como coeficiente no agravamento de pacientes acometidos por COVID-19. Res Soc Dev. 2020;9(9):e395997304.
28. Palaiodimos L, Kokkinidis DG, Li W, Karamanis D, Ognibene J, Arora S, et al. Severe obesity is associated with higher in-hospital mortality in a cohort of patients with COVID-19 in the Bronx, New York. Metabolism [Internet]. 2020;108:154262. Disponível em: https://doi.org/10.1016/j.metabol.2020.154262
29. Apovian CM. Obesity: definition, comorbidities, causes, and burden. Am J Manag Care. 2016;22(7):s176-85.
30. States U, December M, Kompaniyets L, Goodman AB, Belay B, Freedman DS, et al. Body Mass Index and Risk for COVID-19 - Related Hospitalization, Intensive Care Unit Admission, Invasive Mechanical Ventilation, and Death - United States, March - December 2020. MMWR Morb Mortal Wkly Rep. 2021;70:355-61. doi: 10.15585/mmwr.mm7010e4external icon.
31. Codo AC, Davanzo GG, Monteiro LB, de Souza GF, Muraro SP, Virgilio-da-Silva JV, et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1α/Glycolysis-Dependent Axis. Cell Metab. 2020;32(3):437-446.e5.
32. Gheblawi M, Wang K, Viveiros A, Nguyen Q, Zhong JC, Turner AJ, et al. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ Res. 2020;1456-74.
33. Mori J, Oudit GY, Lopaschuk GD. SARS-CoV-2 perturbs the renin-angiotensin system and energy metabolism. Am J Physiol Endocrinol Metab. 2020;319(1):E43-7.
34. Underwood PC, Adler GK. The renin angiotensin aldosterone system and insulin resistance in humans. Curr Hypertens Rep. 2013;15(1):59-70.
35. Aminian A, Tu C. Association of Bariatric Surgery with Clinical Outcomes of SARS-CoV-2 Infection: a Systematic Review and Meta-analysis in the Initial Phase of COVID-19 Pandemic. Obes Surg. 2021 Jan 8:1-7. doi: 10.1007/s11695-020-05213-9.
36. Brandão SCS, Godoi ETAM, Ramos JOX, Melo LMMP, Dompieri LT, Brindeiro Filho DF, et al. The Role of the Endothelium in Severe COVID-19. Arq Bras Cardiol. 2020;115(6):1184-9. doi: 10.36660/abc.20200643.
37. Wang Y, Tikellis C, Thomas MC, Golledge J. Angiotensin converting enzyme 2 and atherosclerosis. Atherosclerosis [Internet]. 2013;226(1):3-8. doi: 10.1016/j.atherosclerosis.2012.08.018.
38. Zhang YH, Zhang YH, Dong XF, Hao QQ, Zhou XM, Yu QT, et al. ACE2 and Ang-(1-7) protect endothelial cell function and prevent early atherosclerosis by inhibiting inflammatory response. Inflamm Res. 2015;64(3-4):253-60. doi: 10.1007/s00011-015-0805-1.
39. Castro-Chaves P, Cerqueira R, Pintalhao M, Leite-Moreira AF. New pathways of the renin-angiotensin system: The role of ACE2 in cardiovascular pathophysiology and therapy. Expert Opin Ther Targets. 2010;14(5):485-96.
40. Zhang C, Zhao YX, Zhang YH, Zhu L, Deng BP, Zhou ZL, et al. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc Natl Acad Sci USA. 2010;107(36):15886-91.
41. Thatcher SE, Zhang X, Howatt DA, Lu H, Gurley SB, Daugherty A, et al. Angiotensin-converting enzyme 2 deficiency in whole body or bone marrow-derived cells increases atherosclerosis in low-density lipoprotein receptor -/- mice. Arterioscler Thromb Vasc Biol. 2011;31(4):758-65.
42. Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, et al. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res. 2010;107(7):888-97.
43. Lovren F, Pan Y, Quan A, Teoh H, Wang G, Shukla PC, et al. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am J Physiol - Hear Circ Physiol. 2008;295(4):1377-84.
44. Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, et al. Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol [Internet]. 2020;7(8):e575-82. doi: 10.1016/S2352-3026(20)30216-7.
45. Akbar AN, Gilroy DW. Aging immunity may exacerbate COVID-19. Science. 2020;369(6501):256-7. doi: 10.1126/science.abb0762.
46. Guan W, Ni Z, Hu Y, Liang W, Ou C, He J, et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N Engl J Med. 2020;382(18):1708-20.
47. Wang D, Hu B, Hu C, Zhu F, Liu X, Zhang J, et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA. 2020;323(11):1061-9. doi: 10.1001/jama.2020.1585.
48. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet [Internet]. 2020;395(10229):1054-62. doi: 10.1016/S0140-6736(20)30566-3.
49. COVID-19 e MICI. Igibd [site da Internet]. 2020. Disponível em: https://igibd.it/ .
50. Lippi G, Wong J, Henry BM. Hypertension in patients with coronavirus disease 2019 (COVID-19): A pooled analysis. Polish Arch Intern Med. 2020;130(4):304-9.
51. Schiffrin EL, Flack JM, Ito S, Muntner P, Webb RC. Hypertension and COVID-19. Am J Hypertens. 2020;33(5):373-4.
52. Tadic M, Cuspidi C, Mancia G, Dell'Oro R, Grassi G. COVID-19, hypertension and cardiovascular diseases: Should we change the therapy? Pharmacol Res [Internet]. 2020;158(May):104906. doi: 10.1016/j.phrs.2020.104906.
53. Zhang P, Zhu L, Cai J, Lei F, Qin JJ, Xie J, et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality among Patients with Hypertension Hospitalized with COVID-19. Circ Res. 2020;126(12):1671-81.
54. Zheng YY, Ma YT, Zhang JY, Xie X. COVID-19 and the cardiovascular system. Nat Rev Cardiol [Internet]. 2020;17(5):259-60. doi: 10.1038/s41569-020-0360-5.
55. Chen T, Wu D, Chen H, Yan W, Yang D, Chen G, et al. Clinical characteristics of 113 deceased patients with coronavirus disease 2019: Retrospective study. BMJ [Internet]. 2020;368(December 2019):1-14. doi:10.1136/bmj.m1091.
56. Mennuni S, Rubattu S, Pierelli G, Tocci G, Fofi C, Volpe M. Hypertension and kidneys: Unraveling complex molecular mechanisms underlying hypertensive renal damage. J Hum Hypertens [Internet]. 2014;28(2):74-9. doi: 10.1038/jhh.2013.55.
57. De Batista PR, Palacios R, Martín A, Hernanz R, Médici CT, Silva MASC, et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PLoS One. 2014;9(8).
58. Lopes RD, Macedo AVS, de Barros e Silva PGM, Moll-Bernardes RJ, Feldman A, D'Andréa Saba Arruda G, et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-The BRACE CORONA Trial. Am Heart J. 2020;226:49-59.
59. Lopes RD, Macedo AVS, De Barros e Silva PGM, Moll-Bernardes RJ, Dos Santos TM, Mazza L, et al. Effect of Discontinuing vs Continuing Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers on Days Alive and out of the Hospital in Patients Admitted with COVID-19: A Randomized Clinical Trial. JAMA. 2021;325(3):254-64. doi: 10.1001/jama.2020.25864.
60. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med. 2020;76(April):14-20.
61. Leung JM, Yang CX, Sin DD. COVID-19 and nicotine as a mediator of ACE-2. Eur Respir J. 2020;55(2001261).
62. Russo P, Bonassi S, Giacconi R, Malavolta M, Tomino C, Maggi F. COVID-19 and smoking: Is nicotine the hidden link? Eur Respir J. 2020;55(6):5-9.
63. Farsalinos K, Angelopoulou A, Alexandris N, Poulas K. COVID-19 and the nicotinic cholinergic system. Eur Respir J. 2020;56(1):2001589. doi: 10.1183/13993003.01589-2020.
64. Andersson J. The inflammatory reflex - Introduction. J Intern Med. 2005;257(2):122-5.
65. Kalamida D, Poulas K, Avramopoulou V, Fostieri E, Lagoumintzis G, Lazaridis K, et al. Muscle and neuronal nicotinic acetylcholine receptors: Structure, function and pathogenicity. FEBS J. 2007;274(15):3799-845.
66. Rojas M, Acosta-Ampudia Y, Monsalve DM, Ramírez-Santana C, Anaya JM. How Important Is the Assessment of Soluble ACE-2 in COVID-19? Am J Hypertens. 2021;34(3):296-297. doi: 10.1093/ajh/hpaa178.
67. Batlle D, Wysocki J, Satchell K. Soluble angiotensin-converting enzyme 2: A potential approach for coronavirus infection therapy? Clin Sci. 2020;134(5):543-5.
68. Guo J, Huang Z, Lin L, Lv J. Coronavirus Disease 2019 (COVID-19) and Cardiovascular Disease: A Viewpoint on the Potential Influence of Angiotensin-Converting Enzyme Inhibitors/Angiotensin Receptor Blockers on Onset and Severity of Severe Acute Respiratory Syndrome Coronavirus 2 Infection. J Am Heart Assoc. 2020;9(7):e016219.
2025 Associação Brasileira de Alergia e Imunologia
Rua Domingos de Morais, 2187 - 3° andar - Salas 315-317 - Vila Mariana - CEP 04035-000 - São Paulo, SP - Brasil - Fone: (11) 5575.6888